
Current Issue
2026, 17(3): 355-363.
doi: 10.15886/j.cnki.rdswxb.20250083
 Abstract
Abstract FullText HTML
FullText HTML PDF 1105KB
PDF 1105KB
Abstract:
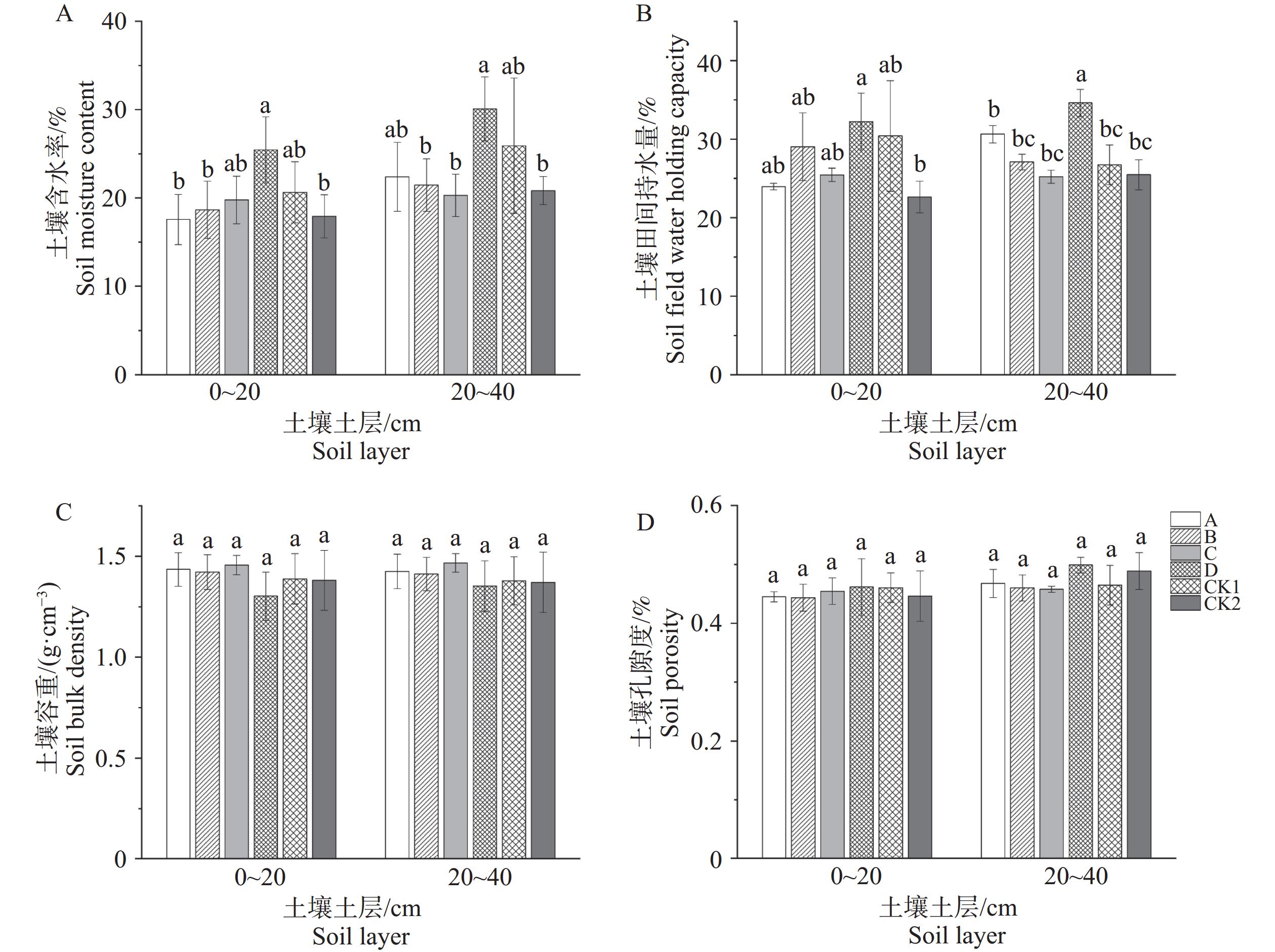
Oil-tea Camellia is a perennial woody oil tree species, and reasonable fertilizer application is of great significance for increasing the oil-tea yield. Compound fertilizer at a reasonable ratio of nitrogen (N), phosphorus (P) and potassium (K) can provide balanced nutrition, which is crucial for improving the growth and yield of oil-tea Camellia. In order to develop a specialized compound fertilizer suitable for the soil conditions of oil-tea Camellia plantations in tropical areas, compound fertilizers at four authoritative ratios of nitrogen, phosphorus and potassium were applied to a plantation of oil-tea Camellia. vietnamensis ‘Houchen 3’, and the leaf nutrient contents, the soil physical and chemical properties, and the enzyme activities in the oil-tea plantation were determined. The results showed that the application of compound fertilizers at different NPK ratios can significantly improve the nutritional elements of oil-tea Camellia leaves, as well as the soil physical and chemical properties and soil enzyme activity in the oil-tea plantation. In terms of improving leaf nutrients, soil nutrients and biological activity, the C (N:P:K = 1:2:2) and D (N:P:K = 10:6:8) treatments largely have the best effect on oil-tea Camellia in the tropical regions, and had the greatest promoting effect especially on the content of available potassium in soil, with an increase of 92.68% and 116.17% in the 0~20 and 20~40 cm soil layers, respectively, compared to the control without fertilization (CK2), which is of great significance for targeted improvement of soil fertility and available potassium content in Hainan oil-tea Camellia plantations. These results provide data for screening specialized compound fertilizers suitable for oil-tea Camellia in the tropical regions, laying a scientific foundation for further research of specialized compound fertilizers for oil-tea Camellia in the tropical regions.
Oil-tea Camellia is a perennial woody oil tree species, and reasonable fertilizer application is of great significance for increasing the oil-tea yield. Compound fertilizer at a reasonable ratio of nitrogen (N), phosphorus (P) and potassium (K) can provide balanced nutrition, which is crucial for improving the growth and yield of oil-tea Camellia. In order to develop a specialized compound fertilizer suitable for the soil conditions of oil-tea Camellia plantations in tropical areas, compound fertilizers at four authoritative ratios of nitrogen, phosphorus and potassium were applied to a plantation of oil-tea Camellia. vietnamensis ‘Houchen 3’, and the leaf nutrient contents, the soil physical and chemical properties, and the enzyme activities in the oil-tea plantation were determined. The results showed that the application of compound fertilizers at different NPK ratios can significantly improve the nutritional elements of oil-tea Camellia leaves, as well as the soil physical and chemical properties and soil enzyme activity in the oil-tea plantation. In terms of improving leaf nutrients, soil nutrients and biological activity, the C (N:P:K = 1:2:2) and D (N:P:K = 10:6:8) treatments largely have the best effect on oil-tea Camellia in the tropical regions, and had the greatest promoting effect especially on the content of available potassium in soil, with an increase of 92.68% and 116.17% in the 0~20 and 20~40 cm soil layers, respectively, compared to the control without fertilization (CK2), which is of great significance for targeted improvement of soil fertility and available potassium content in Hainan oil-tea Camellia plantations. These results provide data for screening specialized compound fertilizers suitable for oil-tea Camellia in the tropical regions, laying a scientific foundation for further research of specialized compound fertilizers for oil-tea Camellia in the tropical regions.
2026, 17(3): 364-378.
doi: 10.15886/j.cnki.rdswxb.20250035
Abstract:
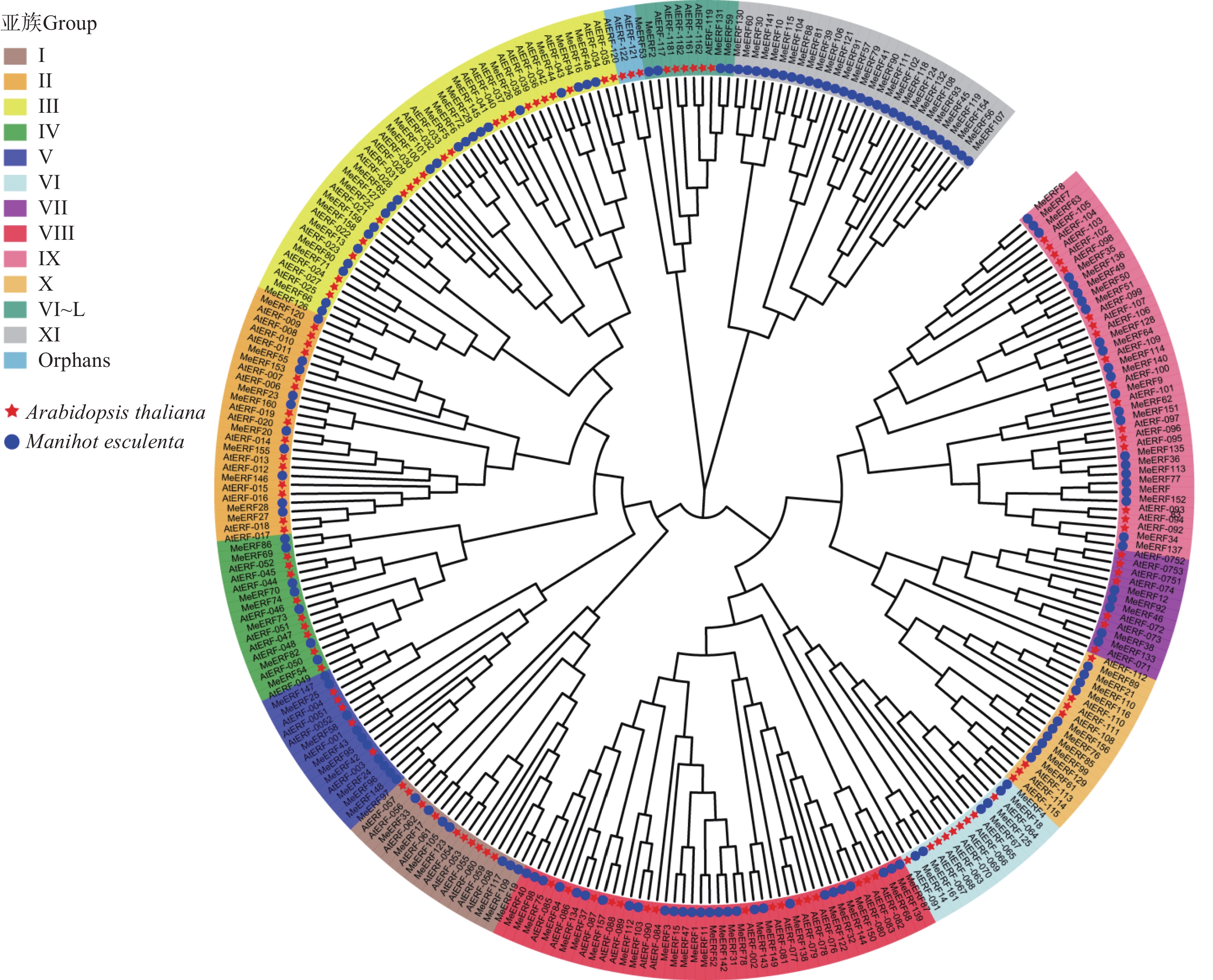
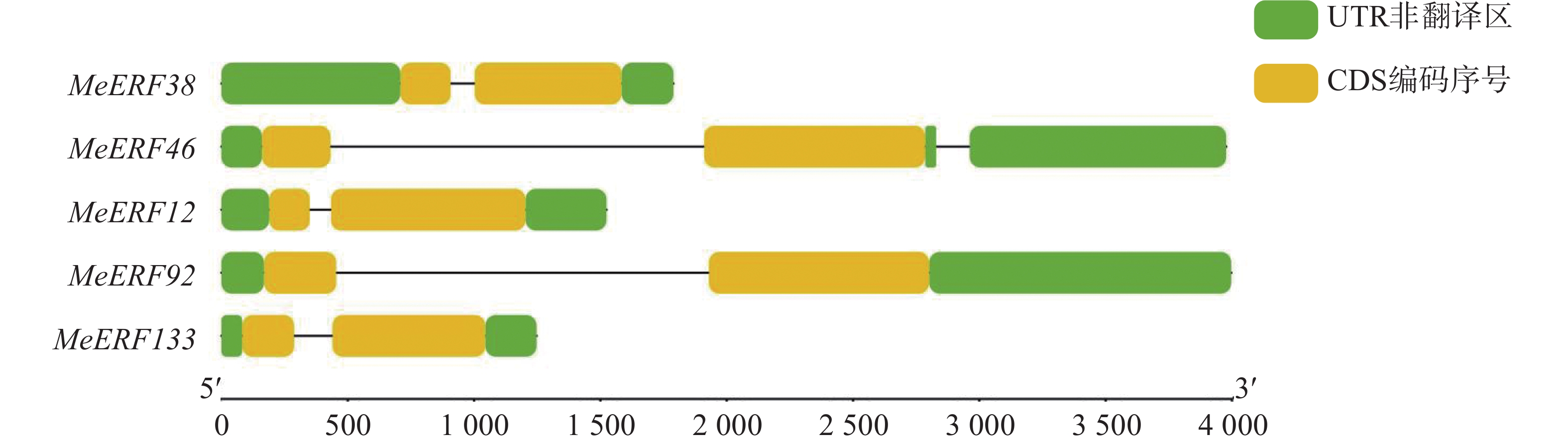
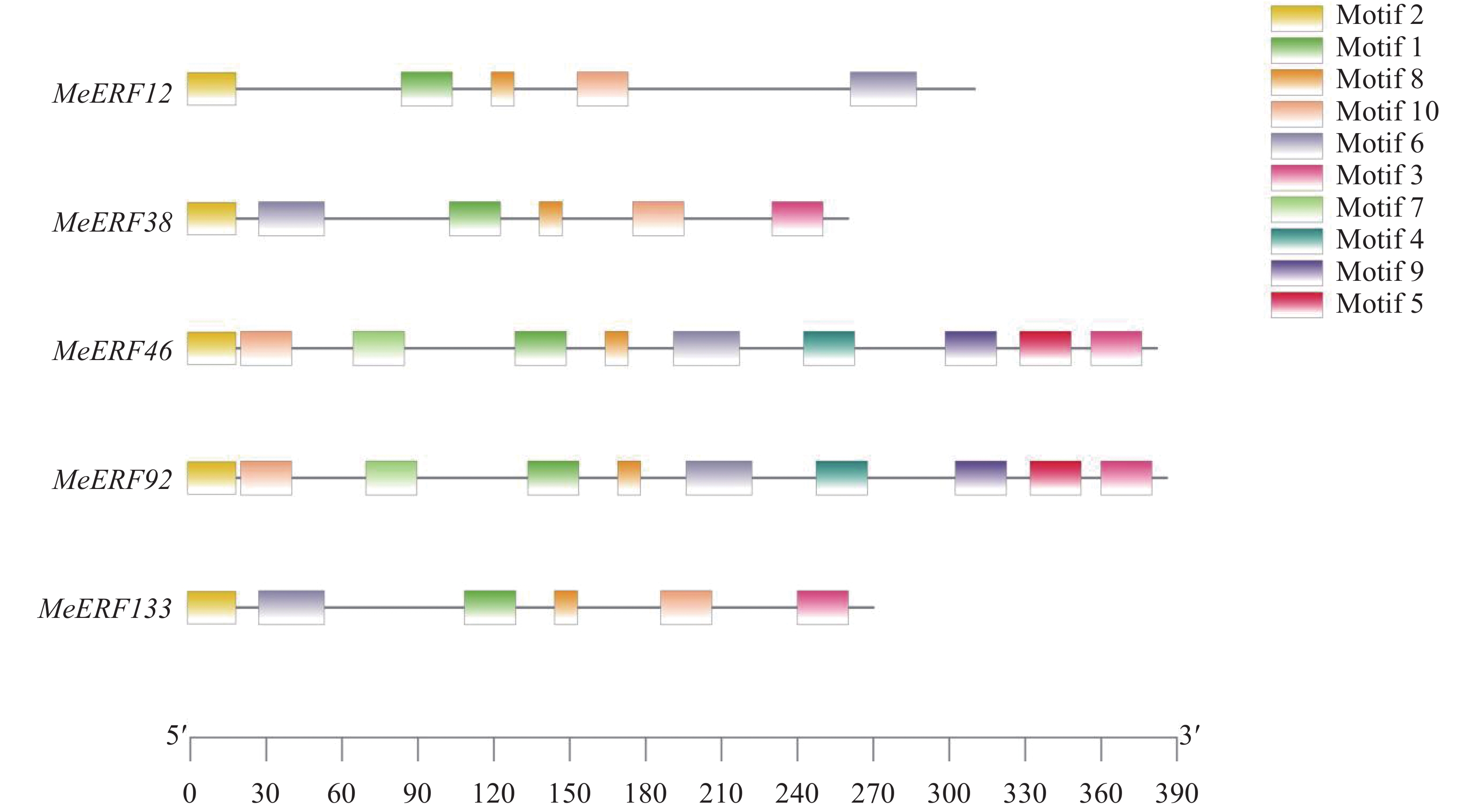
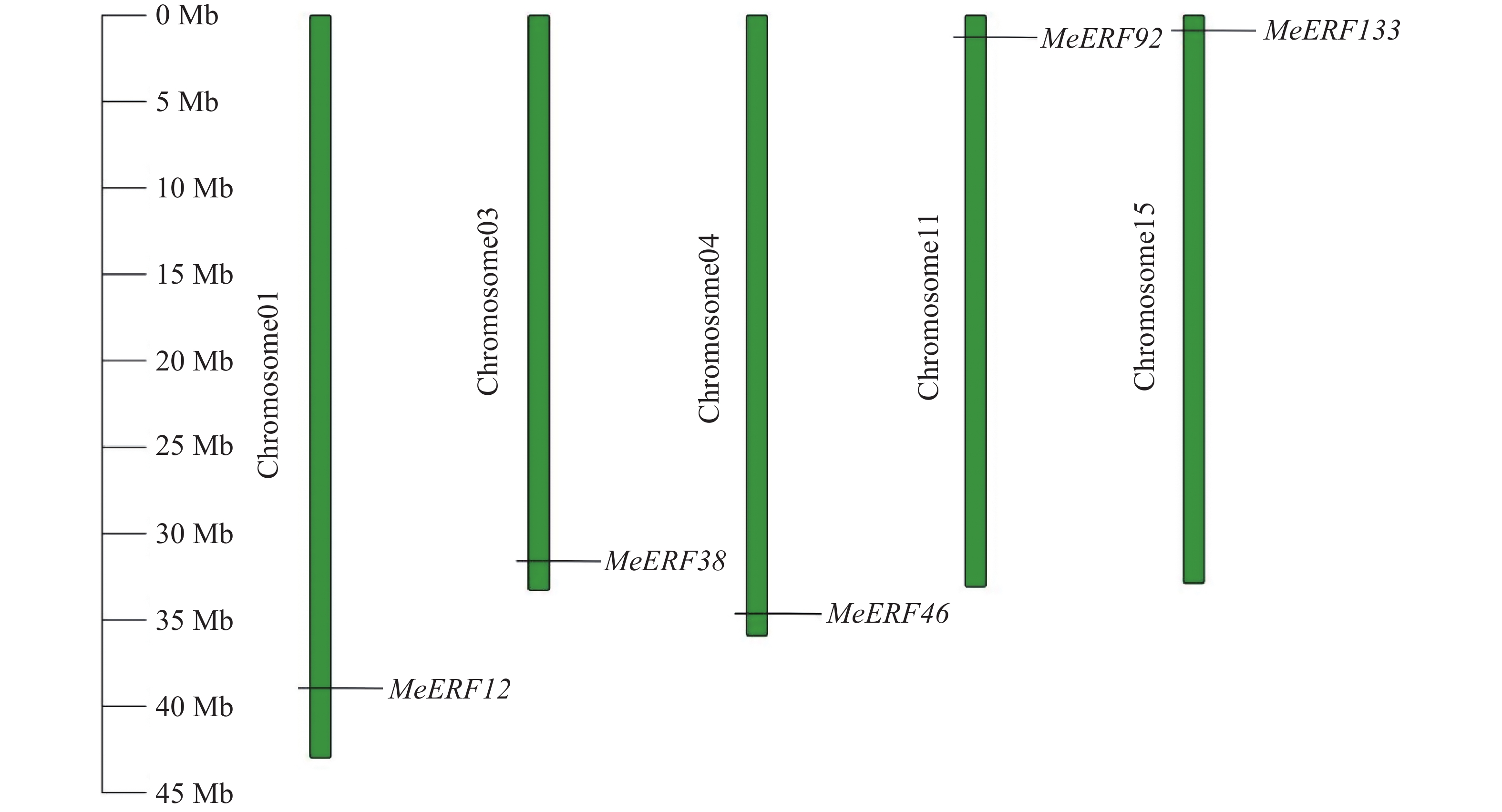
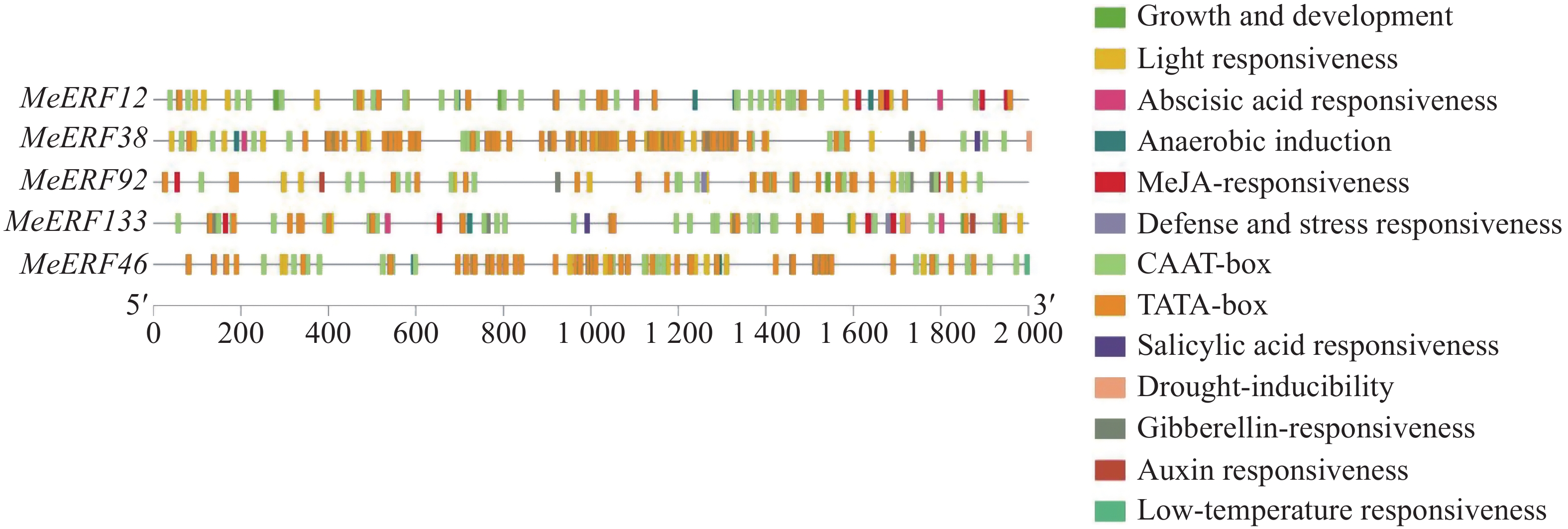
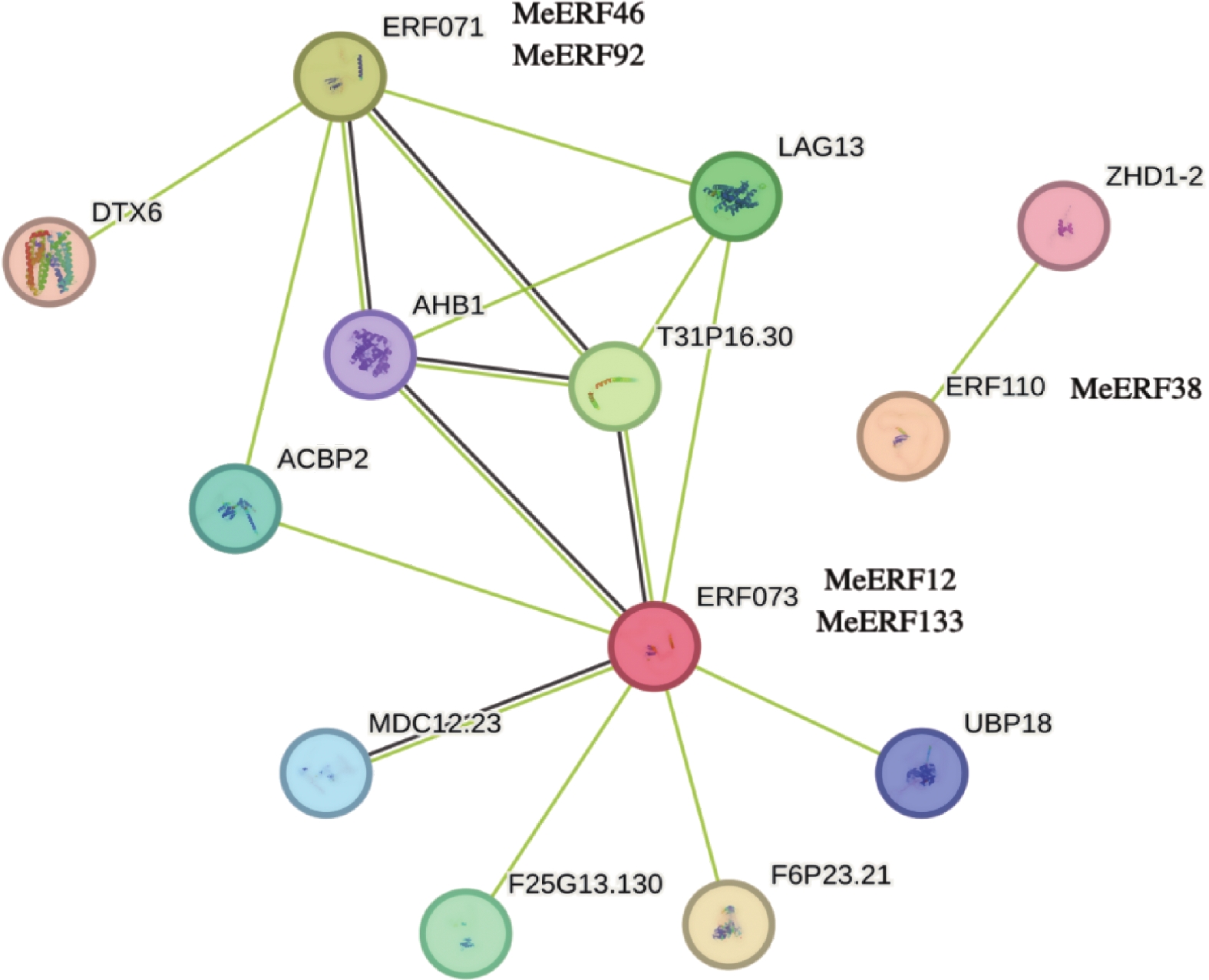
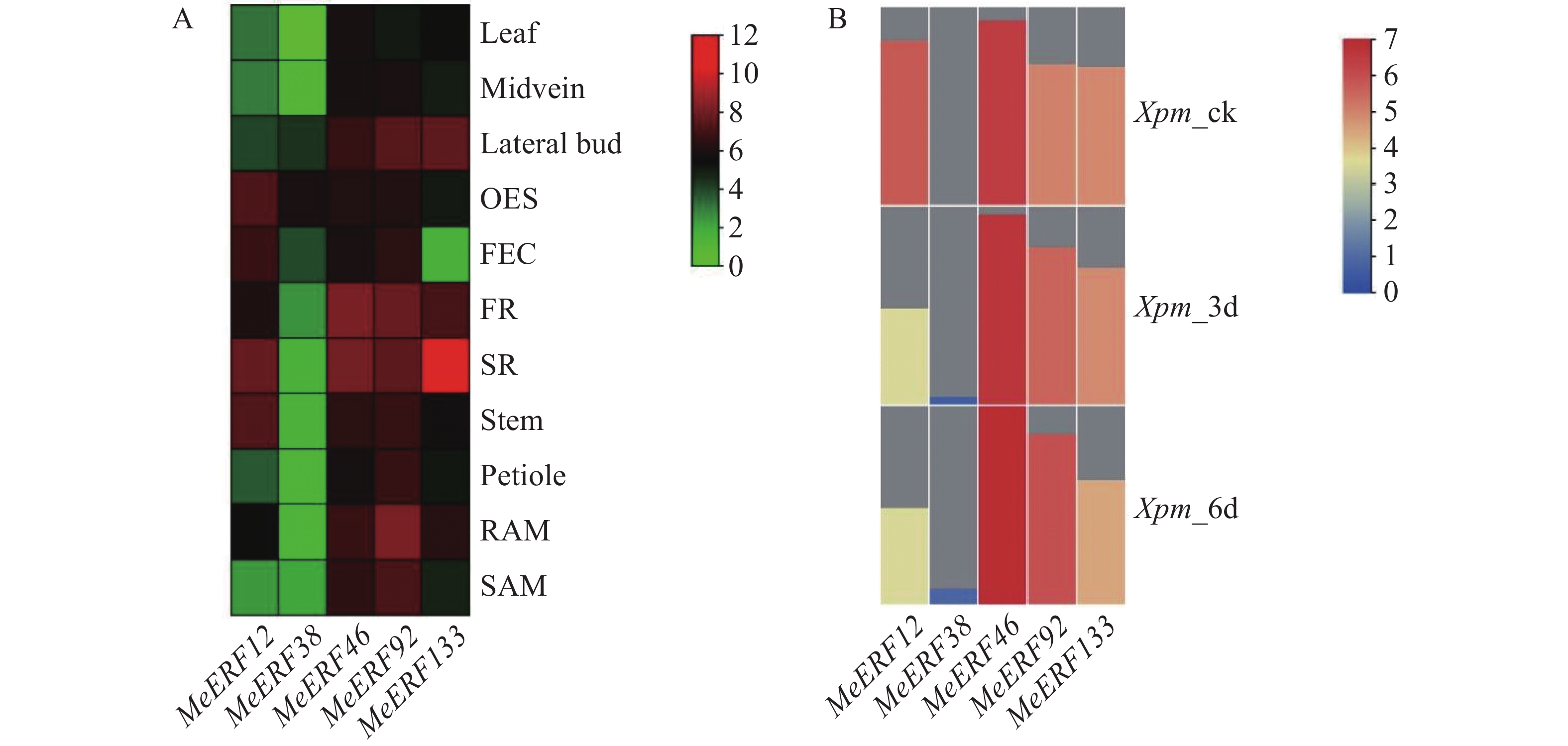
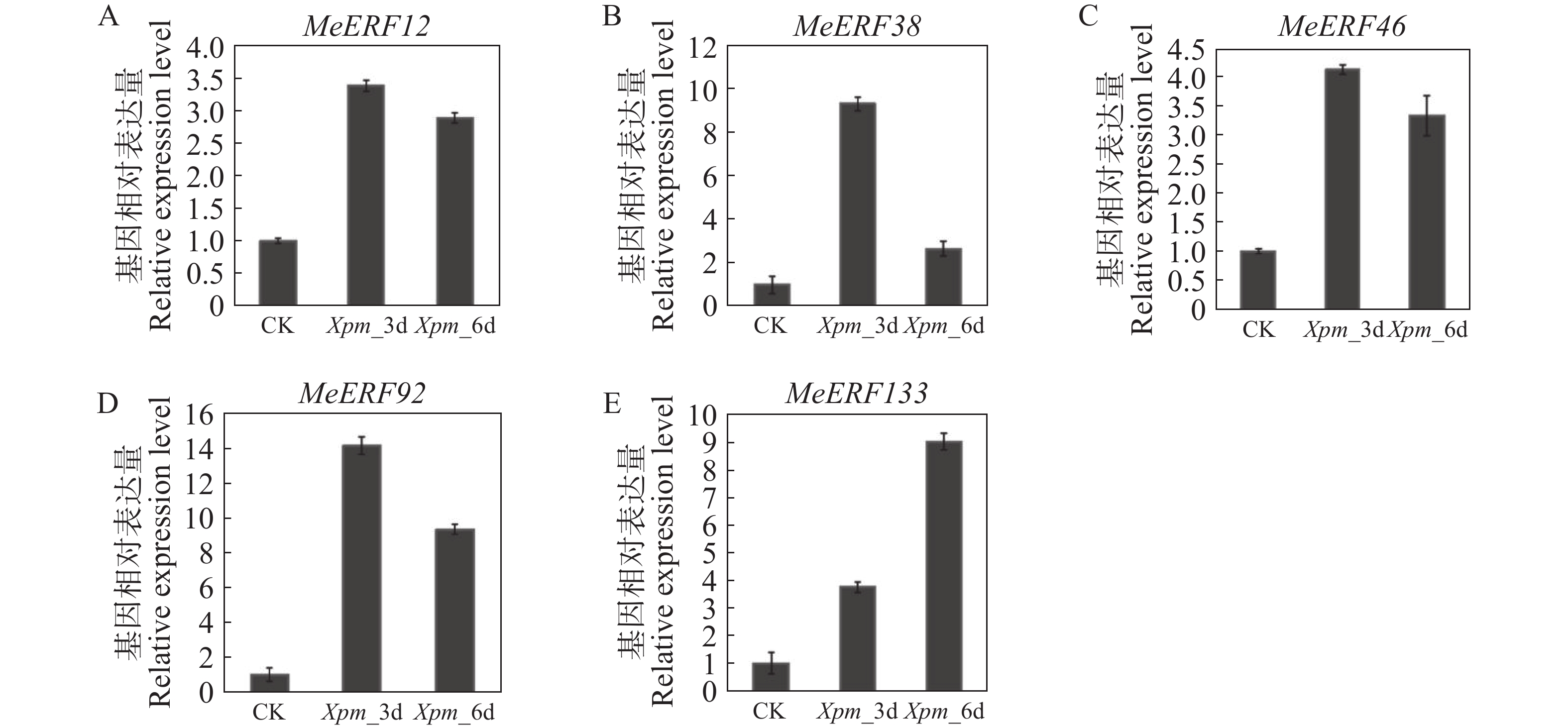
In order to investigate the potential functions of the cassava (Manihot esculenta) Ⅶ subfamily of ERF gene family in response to biotic stress, we identified 161 ERF genes in the cassava genome, which were divided into 13 subfamilies. Analyses were performed on the conserved domains, cis-acting elements in promoter regions, interacting proteins, target genes, and expression patterns of the Ⅶ subfamily members. The results showed that all members of this subfamily contained a conserved AP2 domain, and their promoter regions included 13 types of cis-acting elements related to plant growth and development, and environmental stress responses. Expression pattern analysis revealed that the MeERF46, MeERF133, and MeERF92 genes responded actively to Xpm infection among them, the change of MeERF92 expression was the most significant. WGCNA and protein-protein interaction network analysis indicated that MeERF92 might be involved in the process of oxidative stress, while MeERF133 was widely involved indisease resistance related processes. These findings provide candidate genes for further research into the functions and mechanisms of ERF in cassava's response to biotic stress.
In order to investigate the potential functions of the cassava (Manihot esculenta) Ⅶ subfamily of ERF gene family in response to biotic stress, we identified 161 ERF genes in the cassava genome, which were divided into 13 subfamilies. Analyses were performed on the conserved domains, cis-acting elements in promoter regions, interacting proteins, target genes, and expression patterns of the Ⅶ subfamily members. The results showed that all members of this subfamily contained a conserved AP2 domain, and their promoter regions included 13 types of cis-acting elements related to plant growth and development, and environmental stress responses. Expression pattern analysis revealed that the MeERF46, MeERF133, and MeERF92 genes responded actively to Xpm infection among them, the change of MeERF92 expression was the most significant. WGCNA and protein-protein interaction network analysis indicated that MeERF92 might be involved in the process of oxidative stress, while MeERF133 was widely involved indisease resistance related processes. These findings provide candidate genes for further research into the functions and mechanisms of ERF in cassava's response to biotic stress.
2026, 17(3): 379-389.
doi: 10.15886/j.cnki.rdswxb.20250029
Abstract:
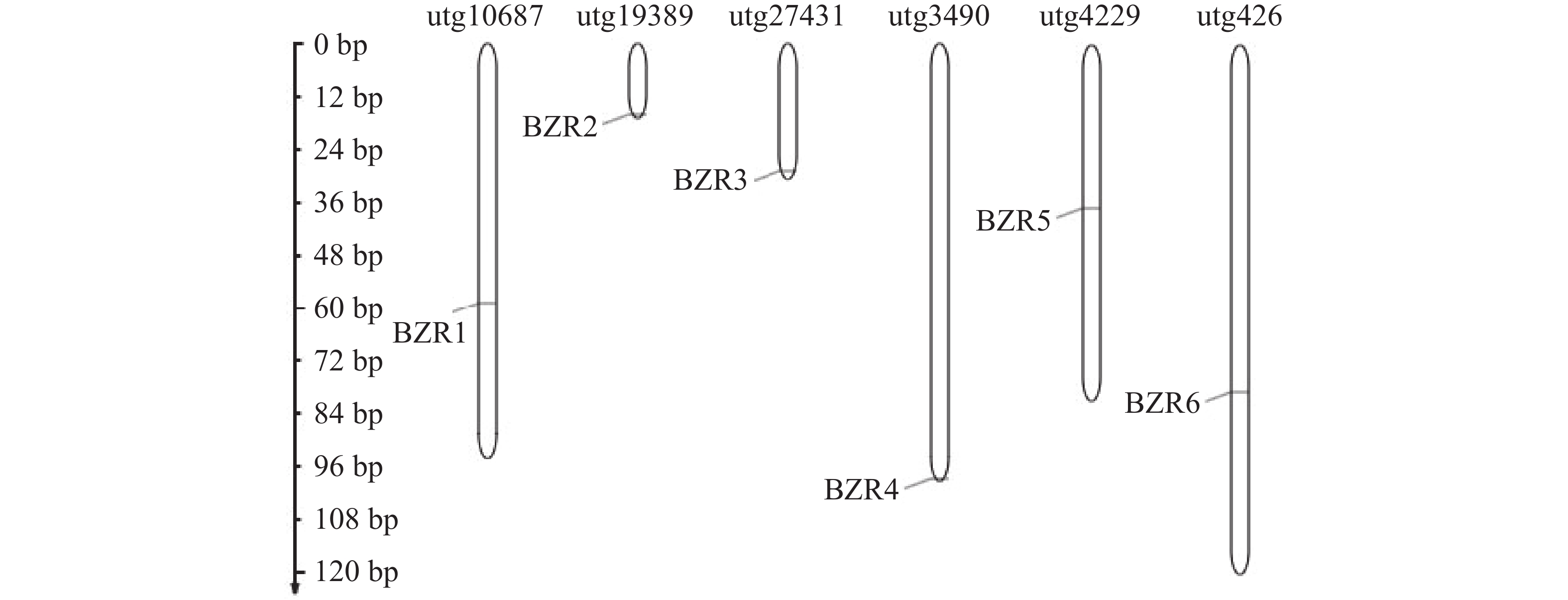
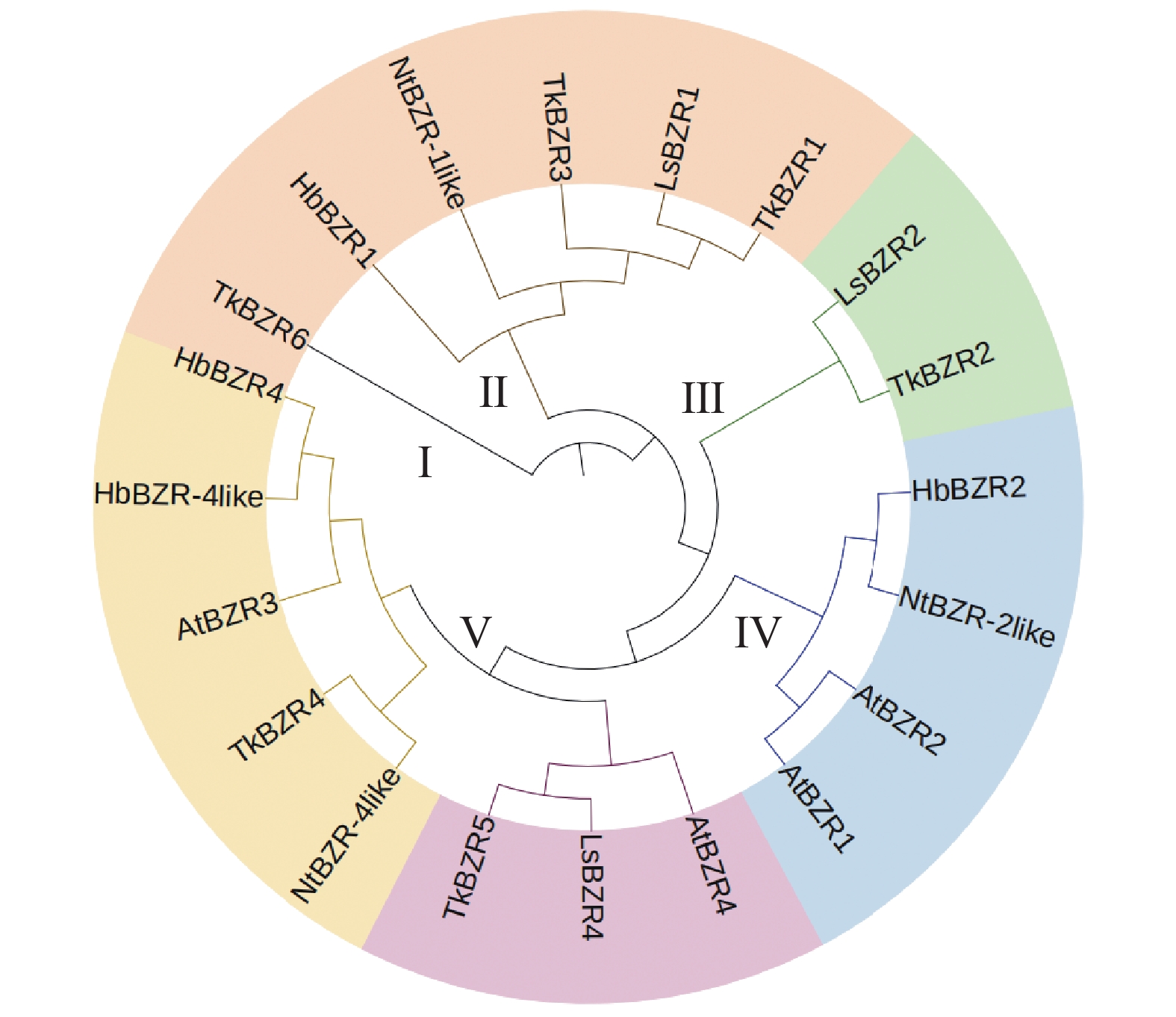
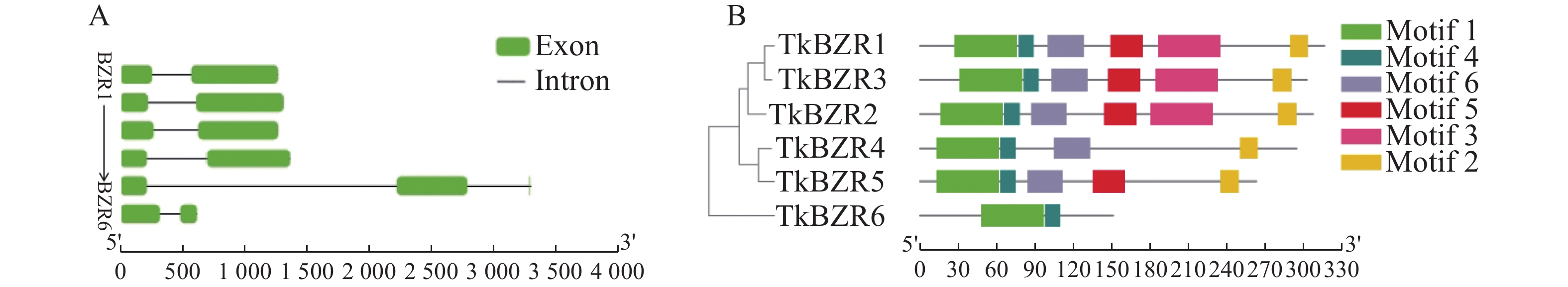
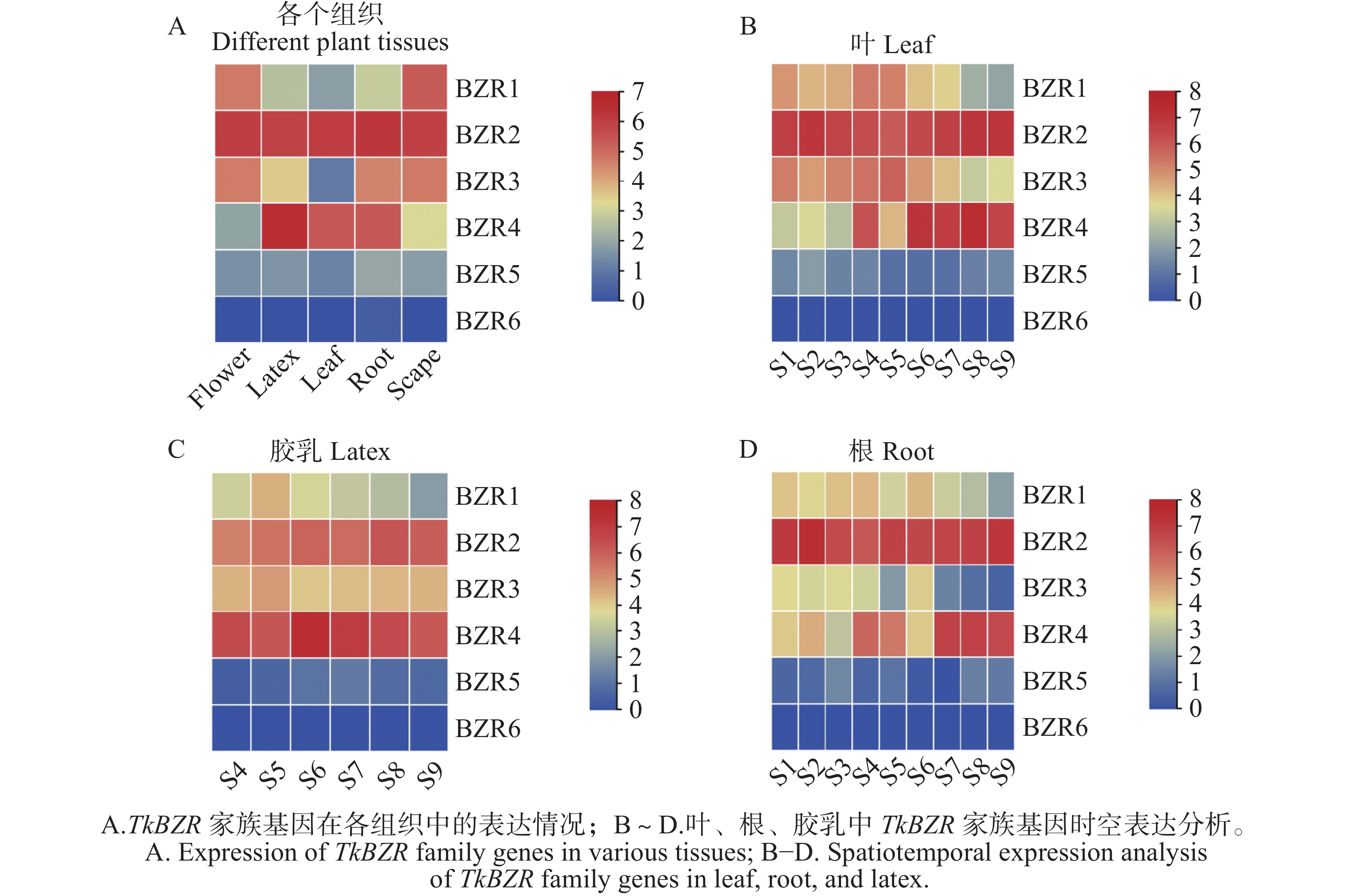
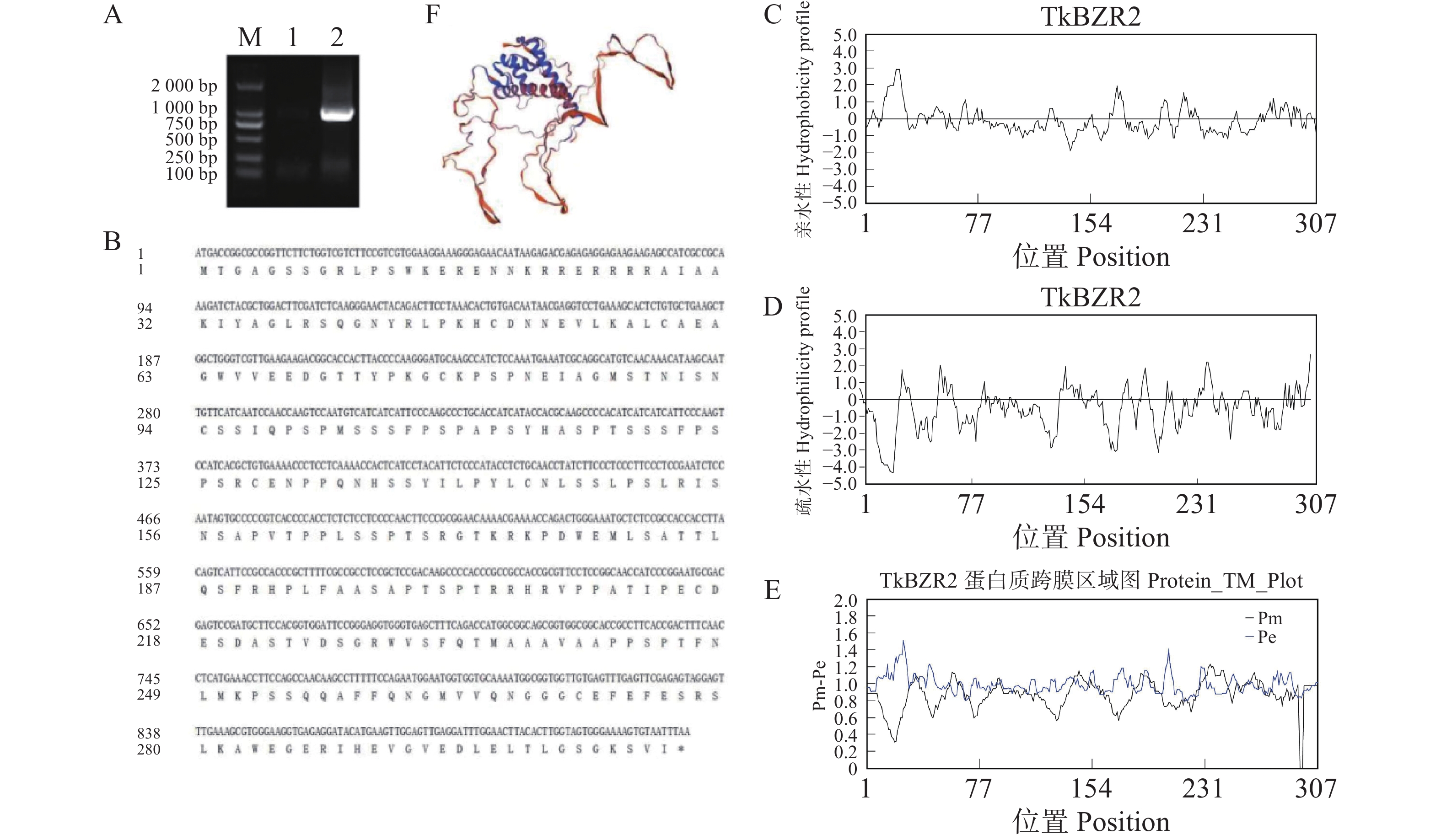
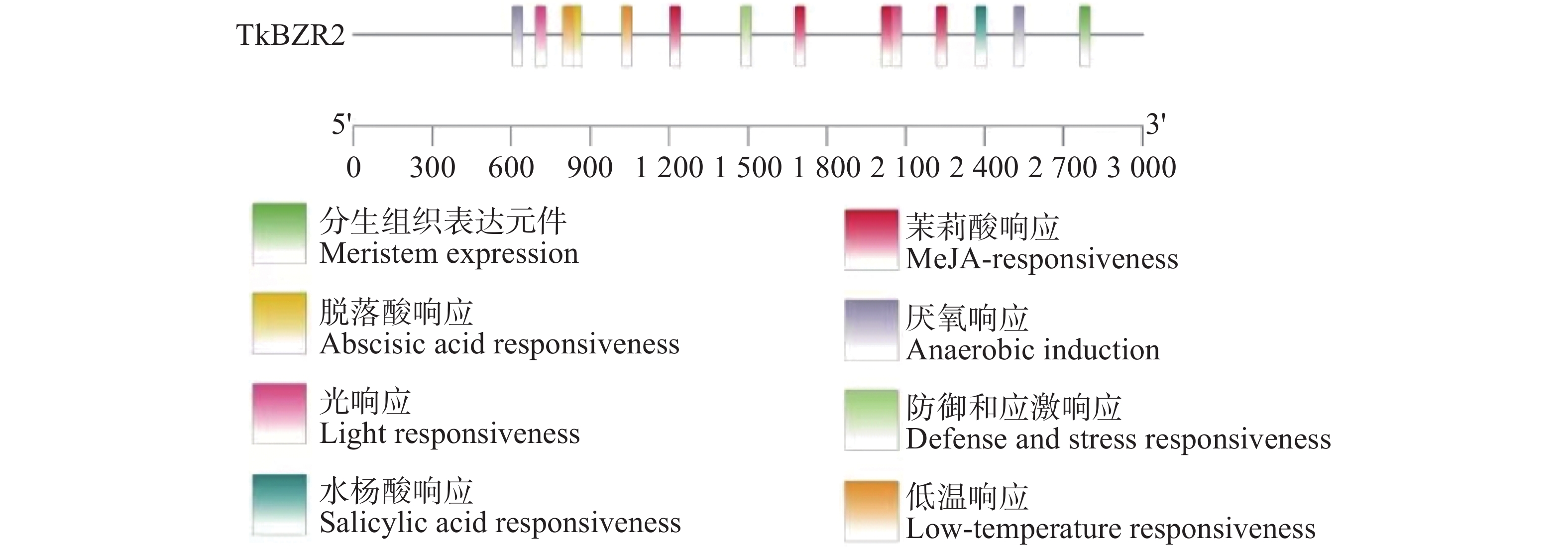
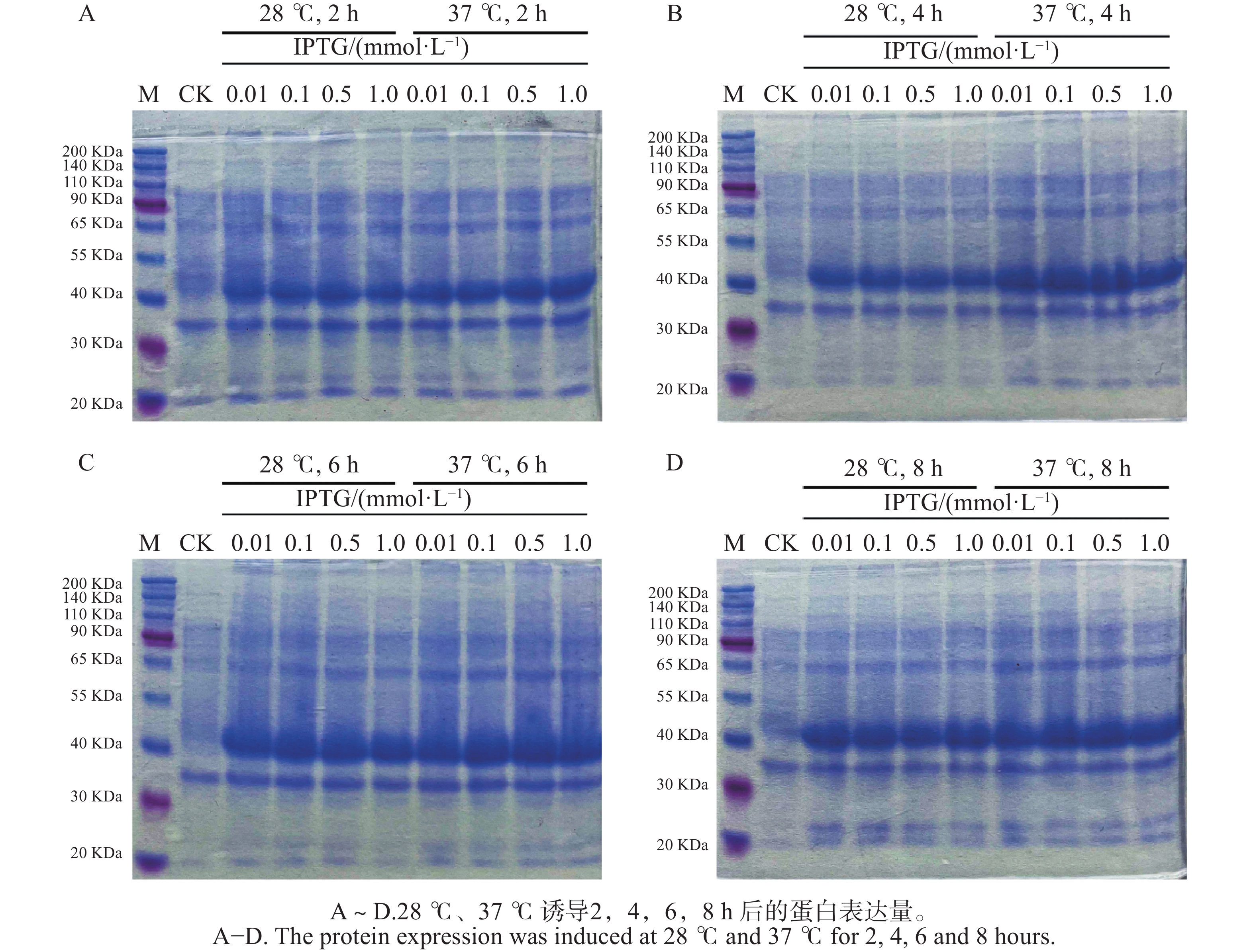
To further understand the role of the TkBZR family genes in the growth, development, and stress resistance of rubber dandelion (Taraxacum kok-saghyz), the TkBZR/BES family genes were identified and analyzed by examining the whole genome sequencing data of T. kok-saghyz. A total of 6 members of the TkBZR family were identified, named from TkBZR1 to TkBZR6, and their distribution on chromosomes, domain characteristics, tissue expression profiles and the spatial-temporal gene expression were analyzed. The results revealed that TkBZR/BES gene family members were distributed on 6 independent scaffolds. Phylogenetic trees indicate that TkBZR/BES gene family members are evolutionarily conserved, with rubber dandelion and lettuce genes clustering in the same subgroup, suggesting a possible close evolutionary relationship between the two. Gene structure and conserved domain analysis show that except for TkBZR5, all the other TkBZR genes contain two exons and one intron, and that all the family members exhibit a highly conserved BES1_N domain. Expression pattern analysis reveals that 5 members are expressed in all five tissues, while one member is almost unexpressed. Additionally, the TkBZR2 gene with the highest expression abundance was successfully cloned, encoding 307 amino acids. Homologous recombination of TkBZR2 was made into prokaryotic expression vector, and the recombinant plasmid was transferred into E. coli BL21 (DE3) .The recombinant protein TkBZR2 was expressed in E. coli BL21 (DE3).
To further understand the role of the TkBZR family genes in the growth, development, and stress resistance of rubber dandelion (Taraxacum kok-saghyz), the TkBZR/BES family genes were identified and analyzed by examining the whole genome sequencing data of T. kok-saghyz. A total of 6 members of the TkBZR family were identified, named from TkBZR1 to TkBZR6, and their distribution on chromosomes, domain characteristics, tissue expression profiles and the spatial-temporal gene expression were analyzed. The results revealed that TkBZR/BES gene family members were distributed on 6 independent scaffolds. Phylogenetic trees indicate that TkBZR/BES gene family members are evolutionarily conserved, with rubber dandelion and lettuce genes clustering in the same subgroup, suggesting a possible close evolutionary relationship between the two. Gene structure and conserved domain analysis show that except for TkBZR5, all the other TkBZR genes contain two exons and one intron, and that all the family members exhibit a highly conserved BES1_N domain. Expression pattern analysis reveals that 5 members are expressed in all five tissues, while one member is almost unexpressed. Additionally, the TkBZR2 gene with the highest expression abundance was successfully cloned, encoding 307 amino acids. Homologous recombination of TkBZR2 was made into prokaryotic expression vector, and the recombinant plasmid was transferred into E. coli BL21 (DE3) .The recombinant protein TkBZR2 was expressed in E. coli BL21 (DE3).
2026, 17(3): 390-398.
doi: 10.15886/j.cnki.rdswxb.20250026
Abstract:
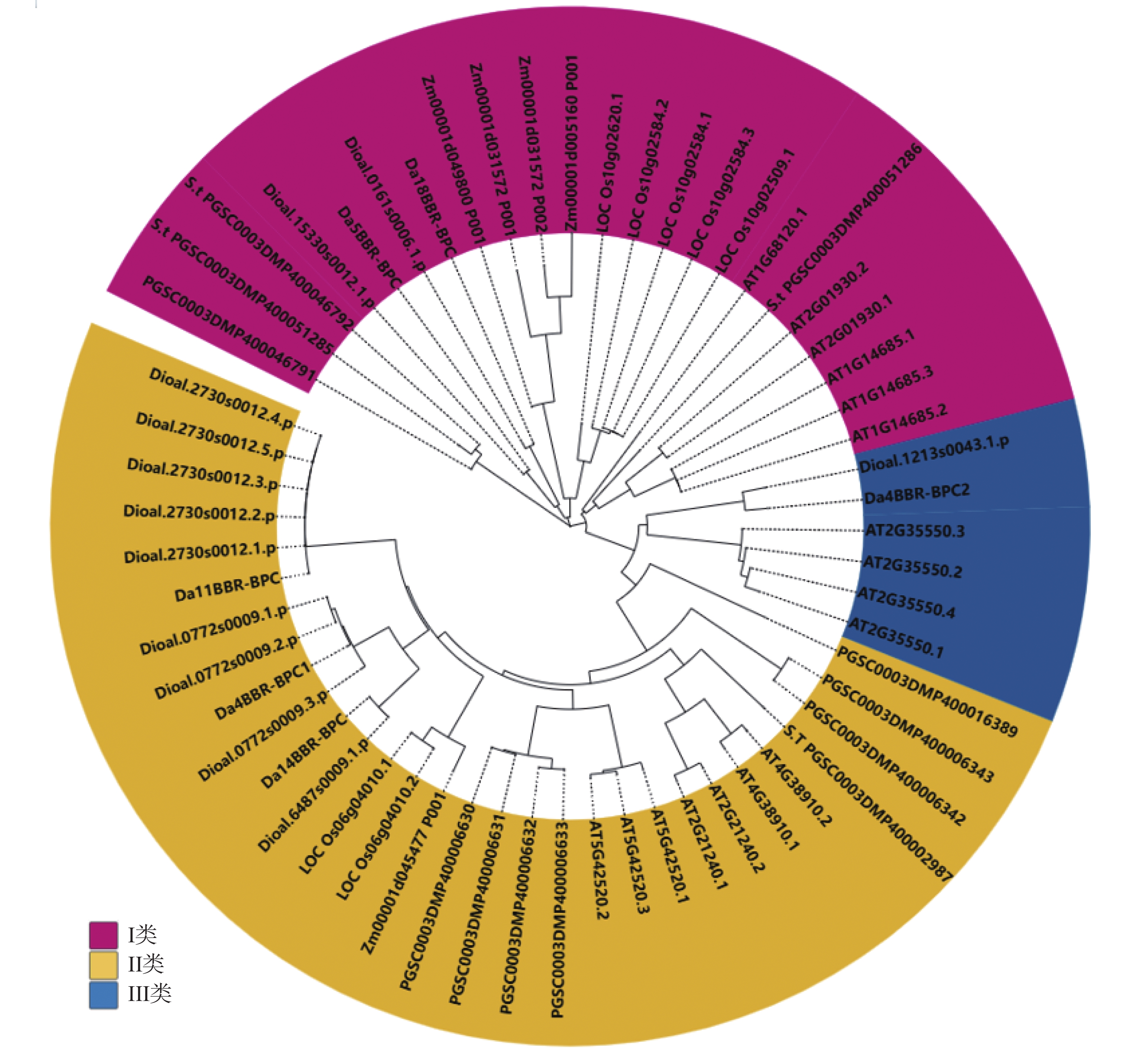
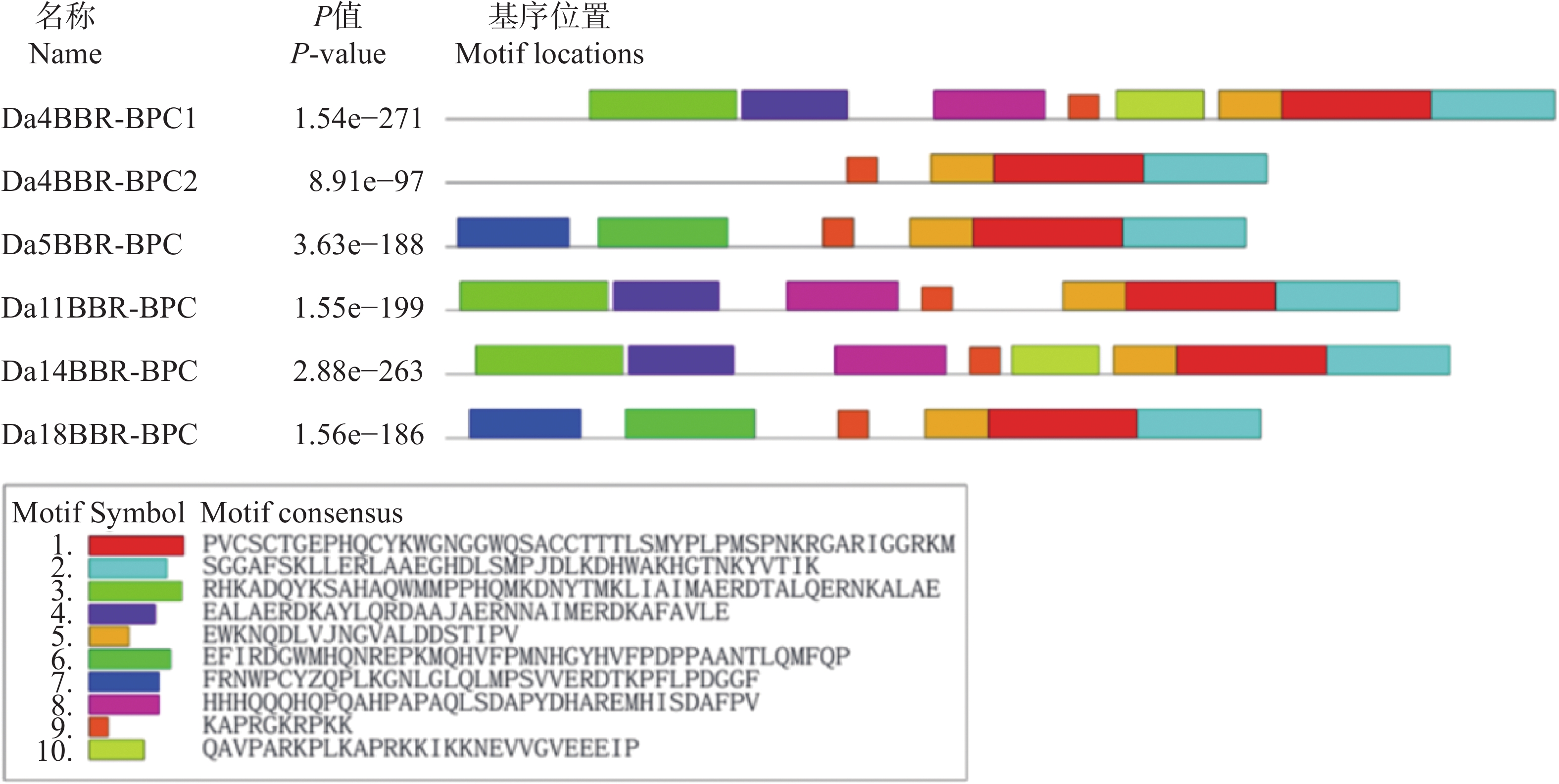
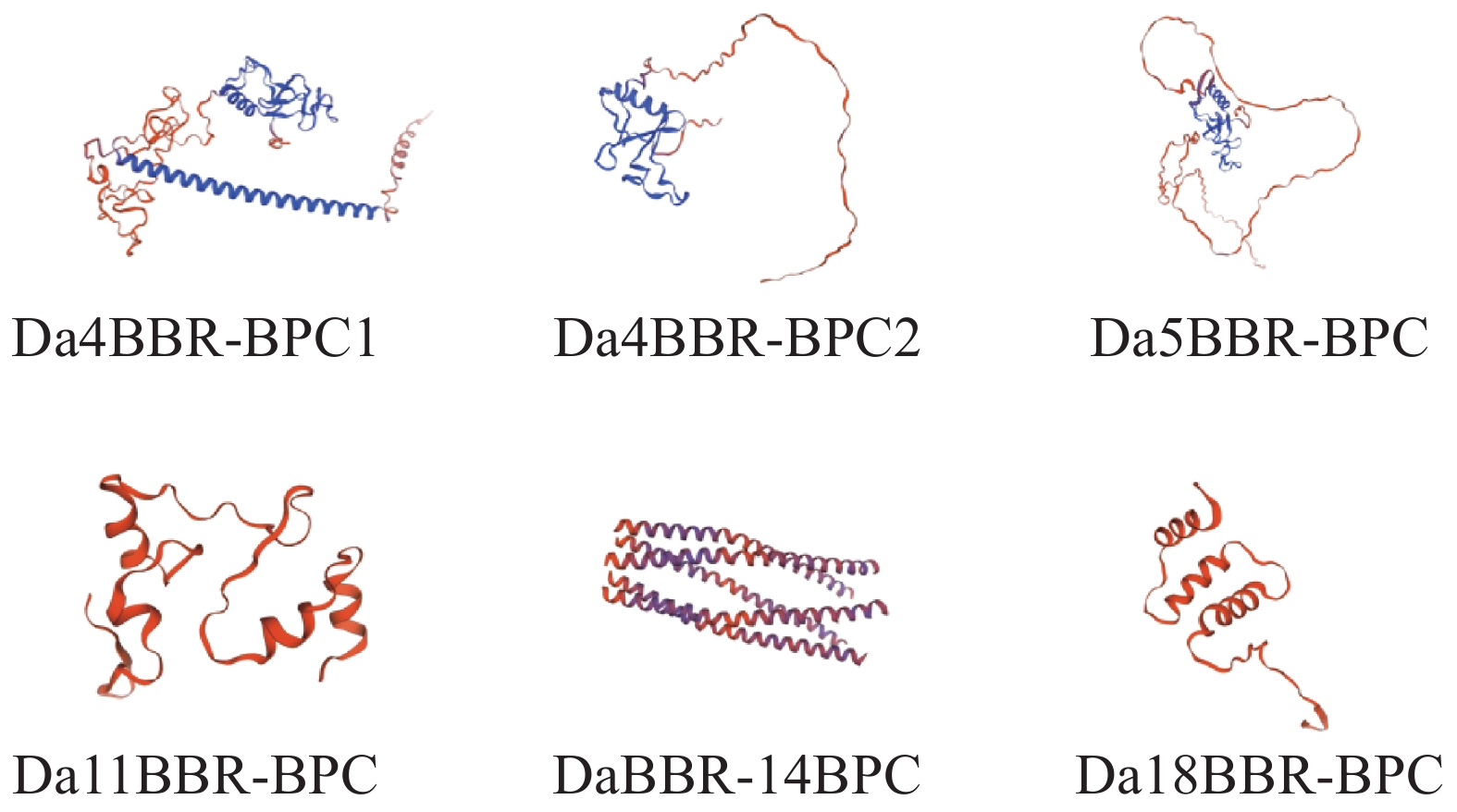
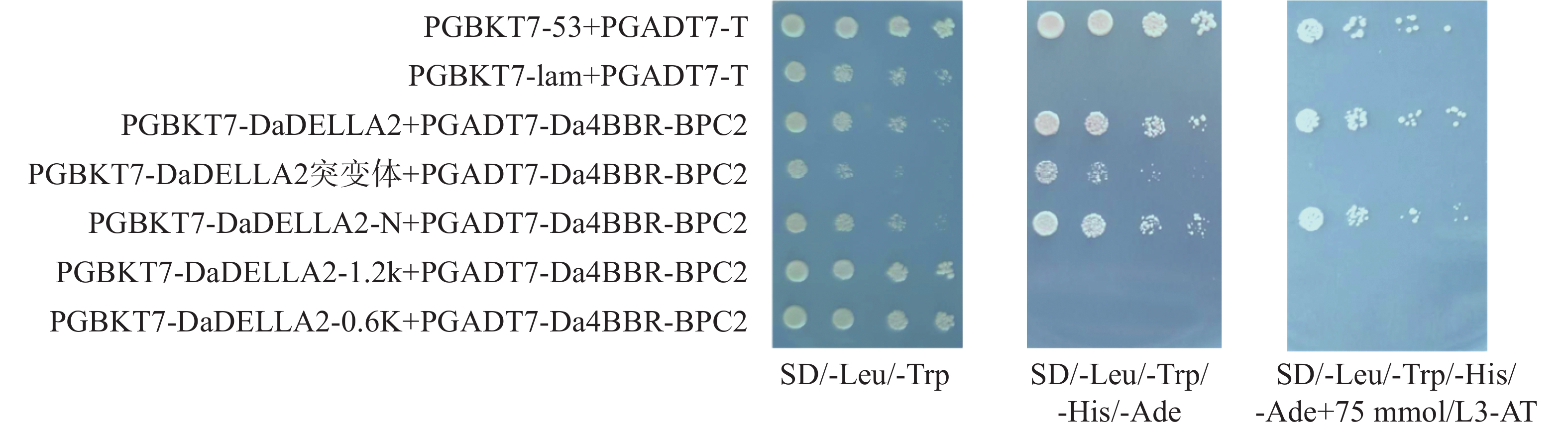
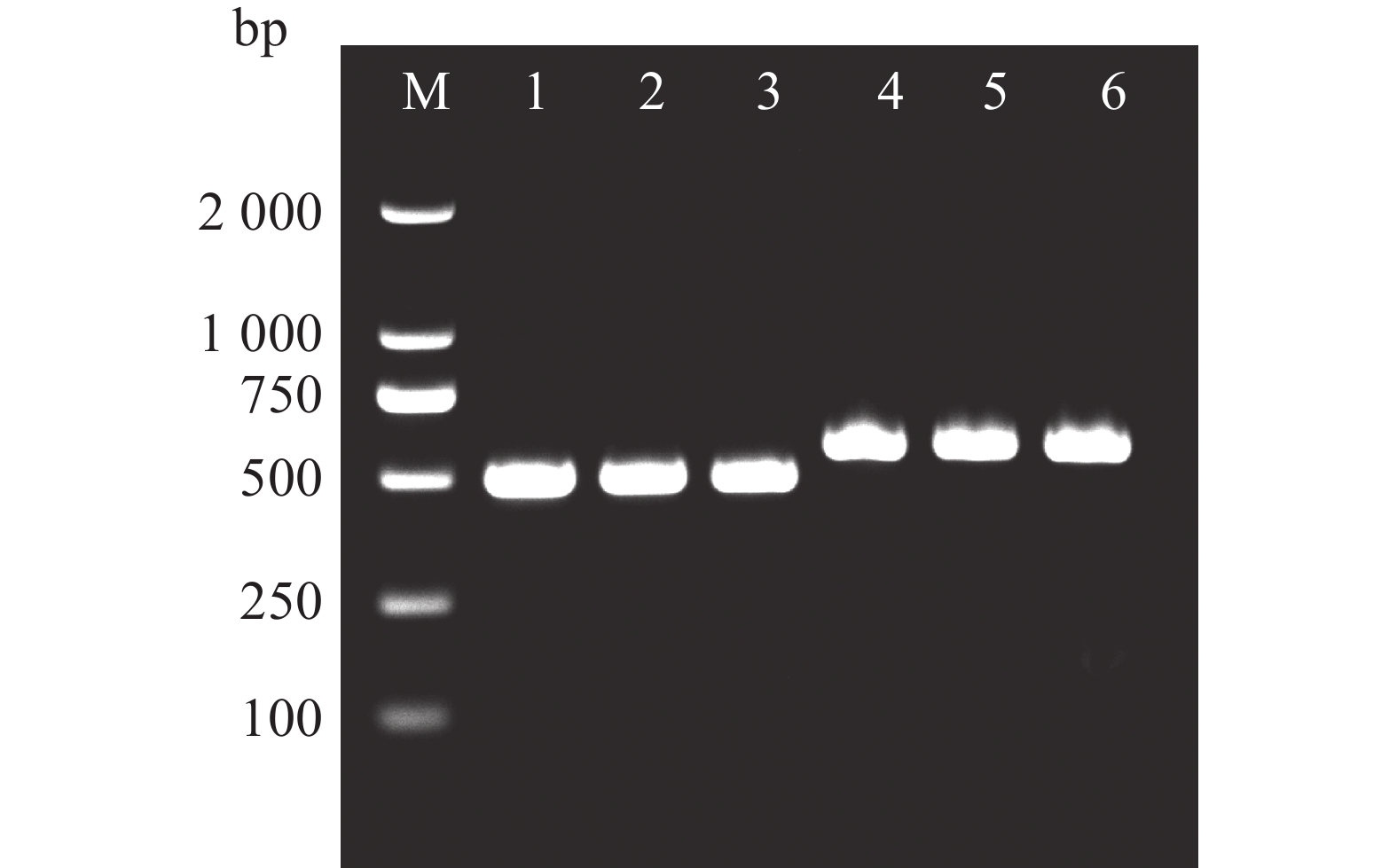
In order to understand the regulatory role of the BBR-BPC gene family in the growth and development of Dioscorea alata (greater yam), we analyzed the gene structure, phylogenetic evolution, conserved motifs, and physicochemical properties of the BBR-BPC gene family in greater yam. We also predicted the secondary and tertiary structures of BBR-BPC proteins and verified the interaction between the BBR-BPC gene and the DELLA gene. Through HMMsearch and BLAST analyses, we identified six members of the BBR-BPC family in the greater yam genome. These gene proteins exhibited significant differences and alternative splicing events, with a total of 10 splice variants detected. Subcellular localization analysis revealed that all members of the BBR-BPC family in greater yam are located in the nucleus.The yeast two-hybrid interaction results between Da4BBR-BPC2 and DaDELLA2 indicate that there is an interaction between Da4BBR-BPC2 and DaDELLA2, occurring at the N-terminus of the DaDELLA2 gene.
In order to understand the regulatory role of the BBR-BPC gene family in the growth and development of Dioscorea alata (greater yam), we analyzed the gene structure, phylogenetic evolution, conserved motifs, and physicochemical properties of the BBR-BPC gene family in greater yam. We also predicted the secondary and tertiary structures of BBR-BPC proteins and verified the interaction between the BBR-BPC gene and the DELLA gene. Through HMMsearch and BLAST analyses, we identified six members of the BBR-BPC family in the greater yam genome. These gene proteins exhibited significant differences and alternative splicing events, with a total of 10 splice variants detected. Subcellular localization analysis revealed that all members of the BBR-BPC family in greater yam are located in the nucleus.The yeast two-hybrid interaction results between Da4BBR-BPC2 and DaDELLA2 indicate that there is an interaction between Da4BBR-BPC2 and DaDELLA2, occurring at the N-terminus of the DaDELLA2 gene.

2026, 17(3): 399-409.
doi: 10.15886/j.cnki.rdswxb.20250015
Abstract:
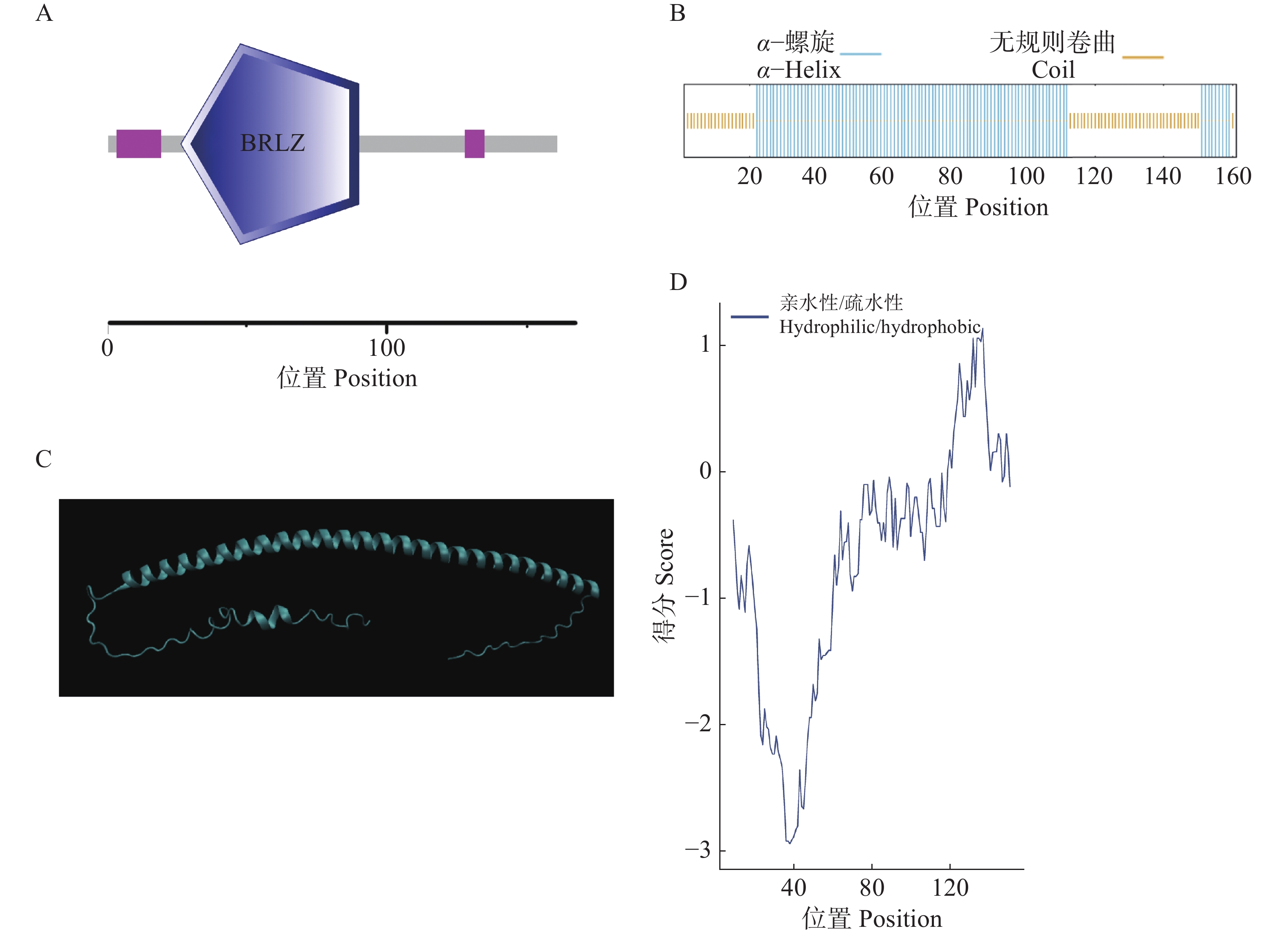
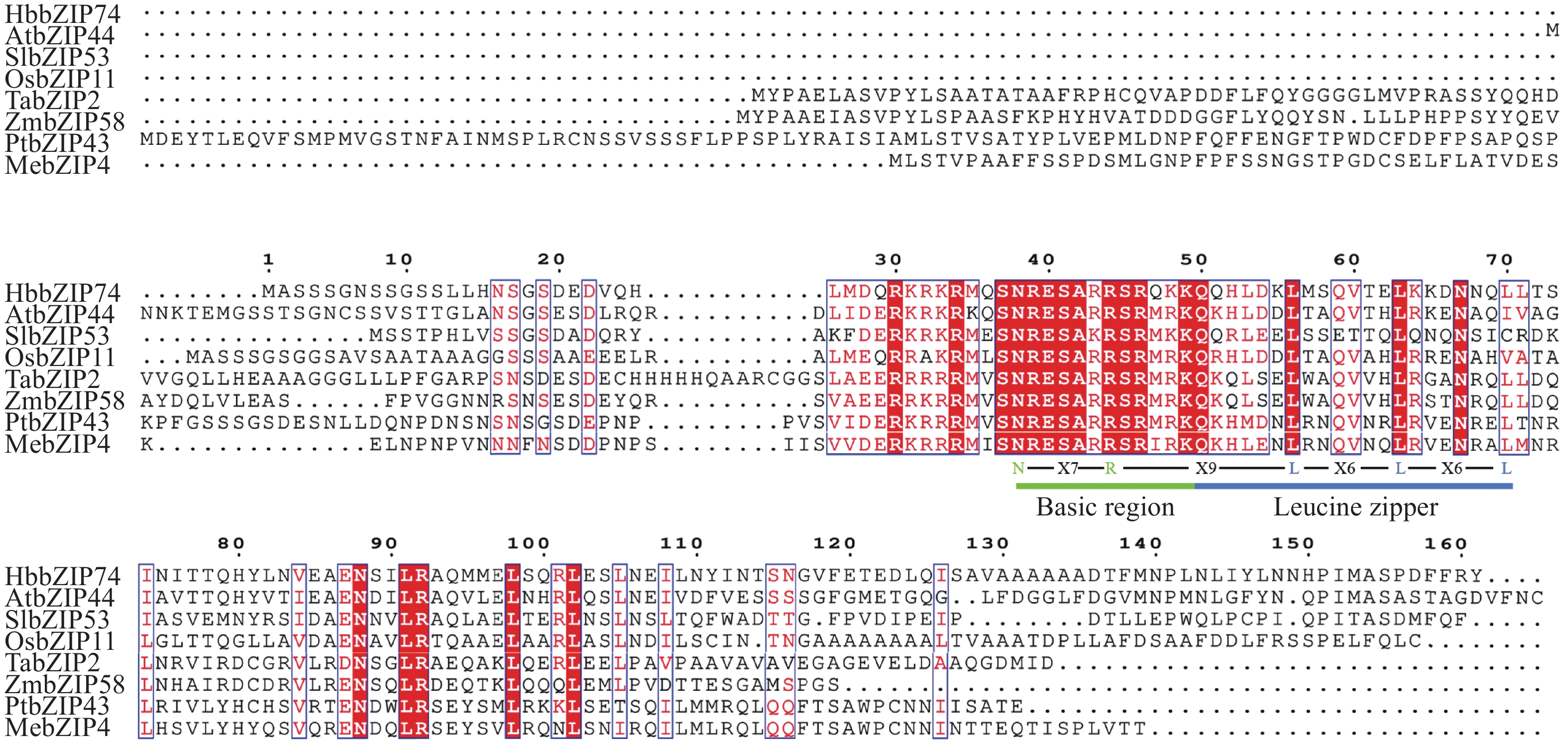
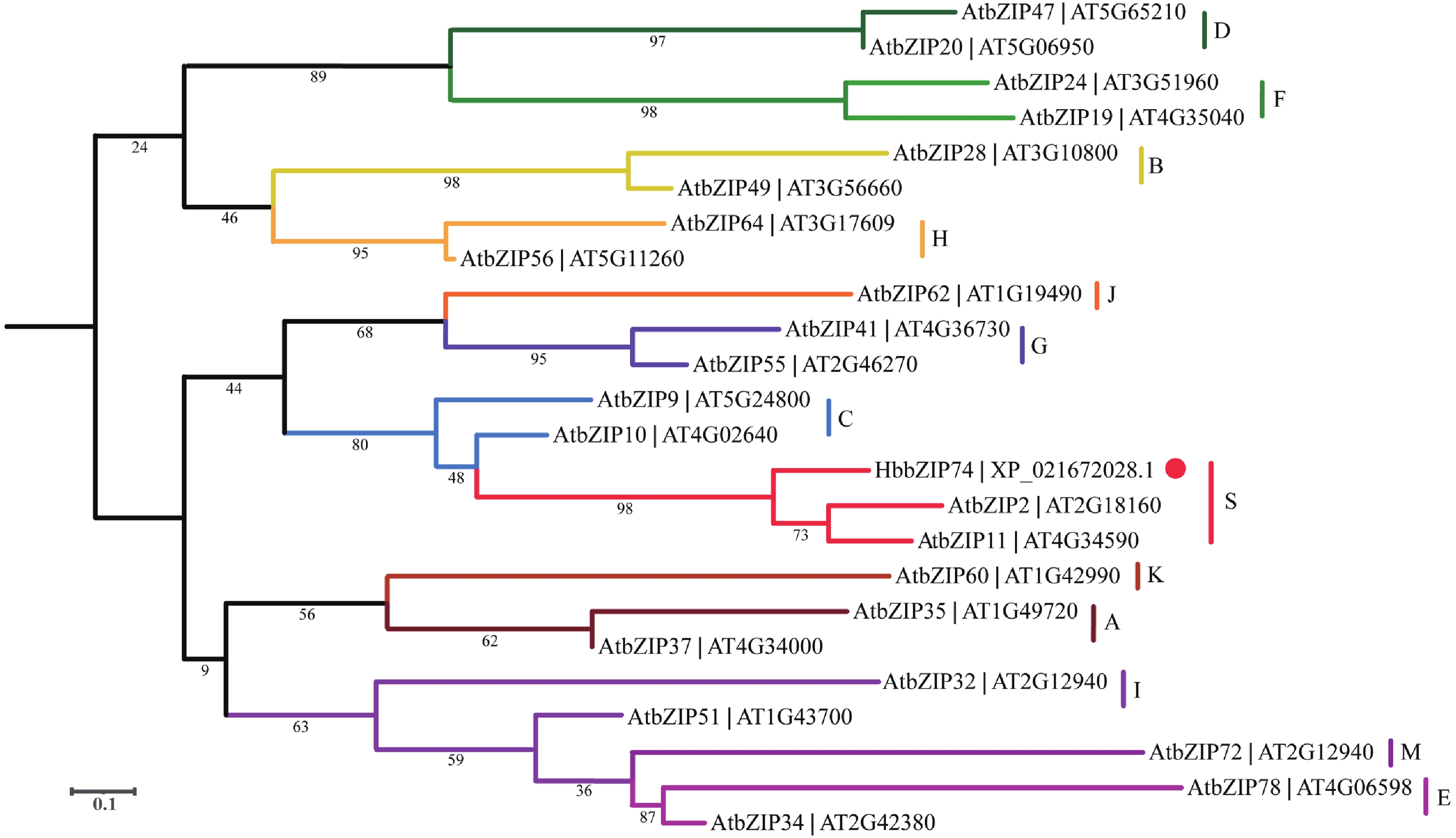
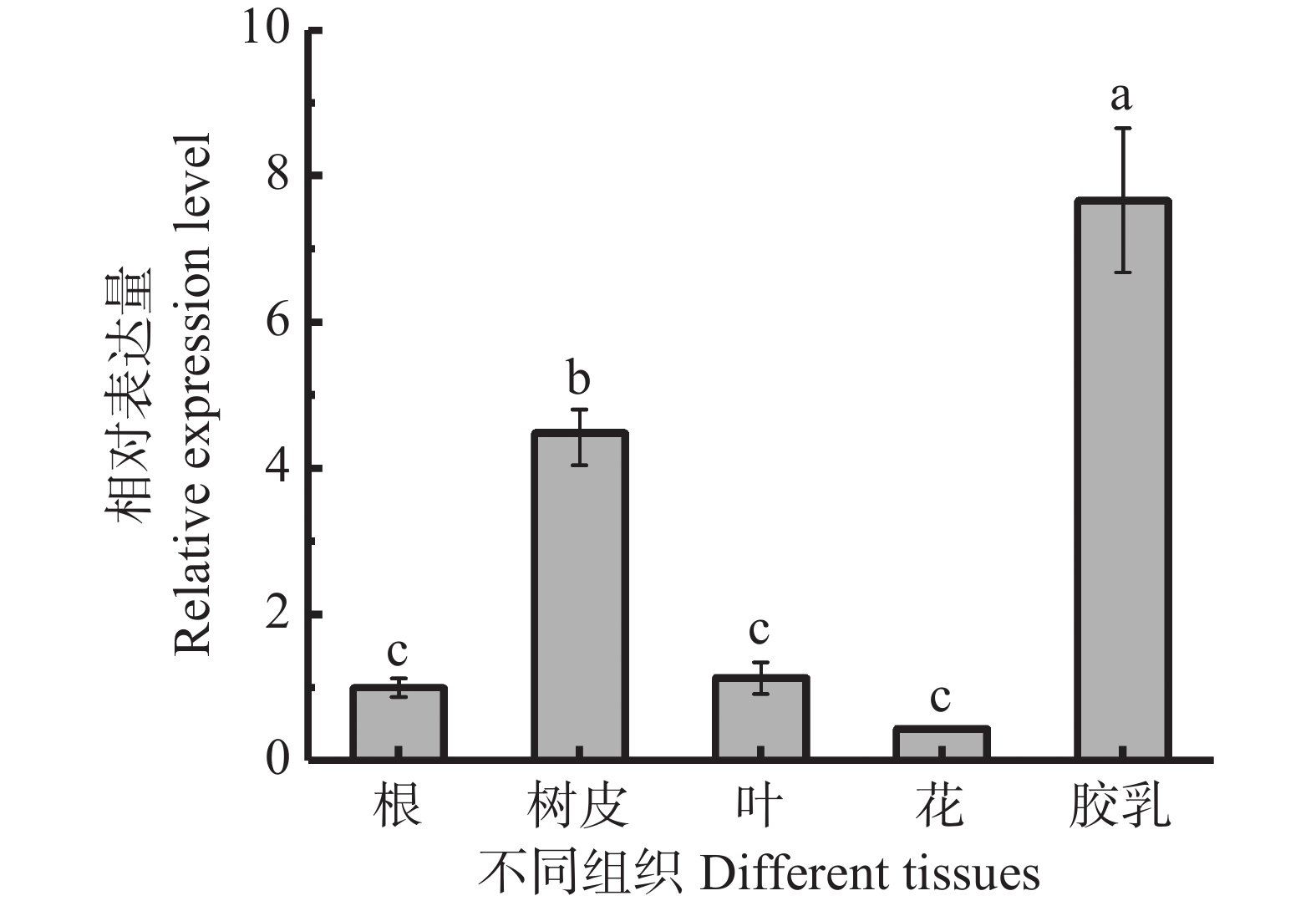
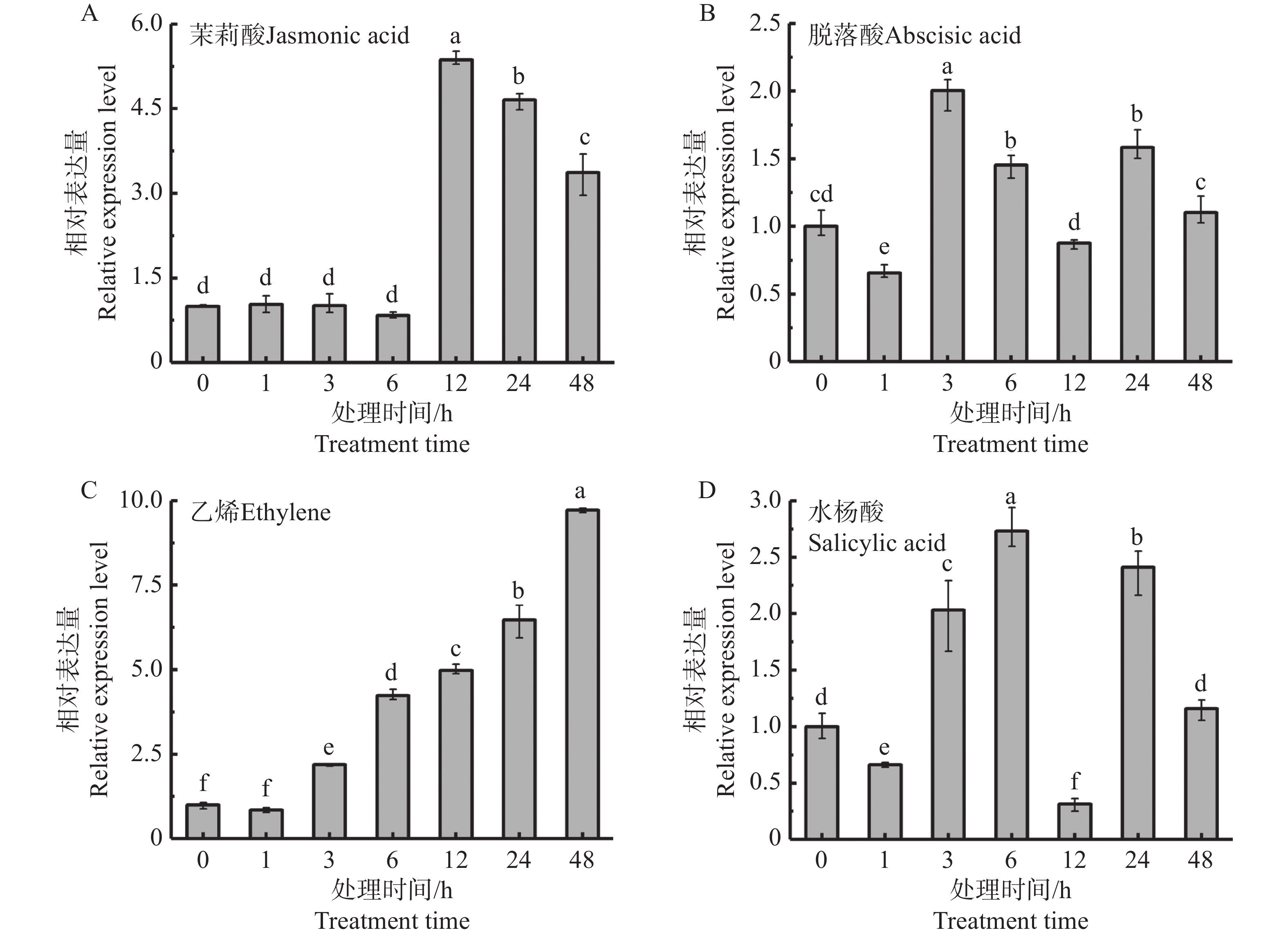
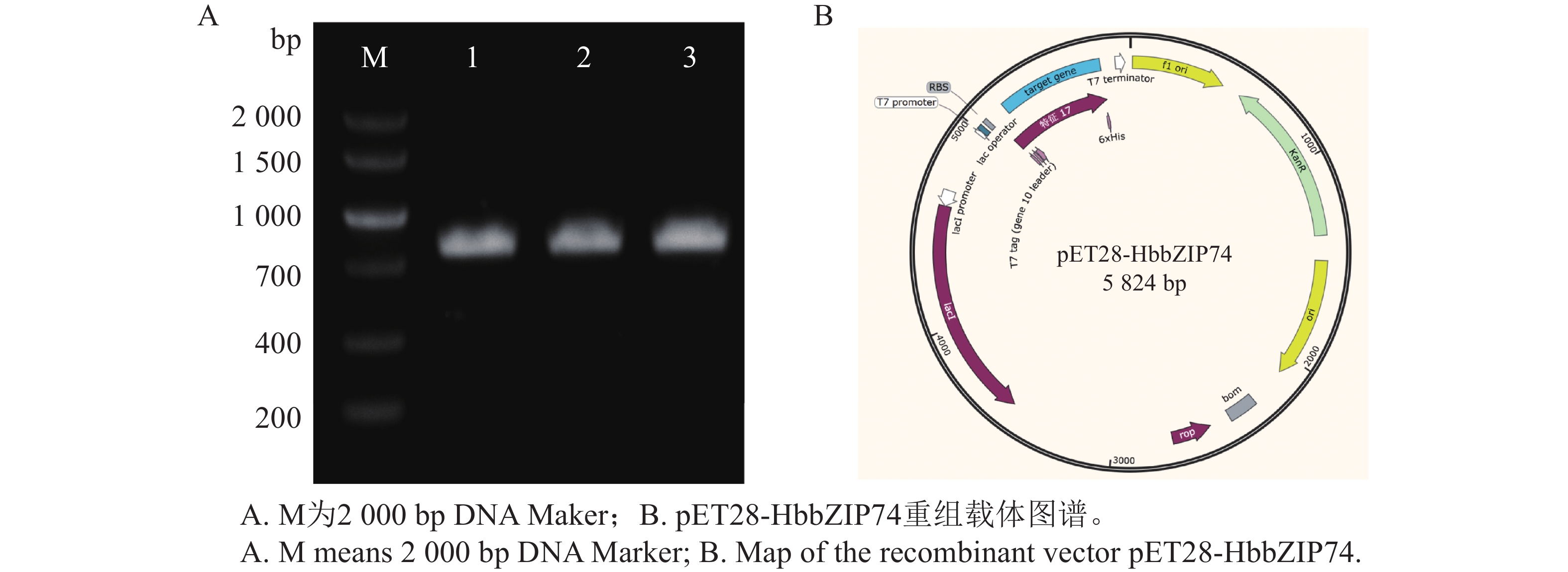
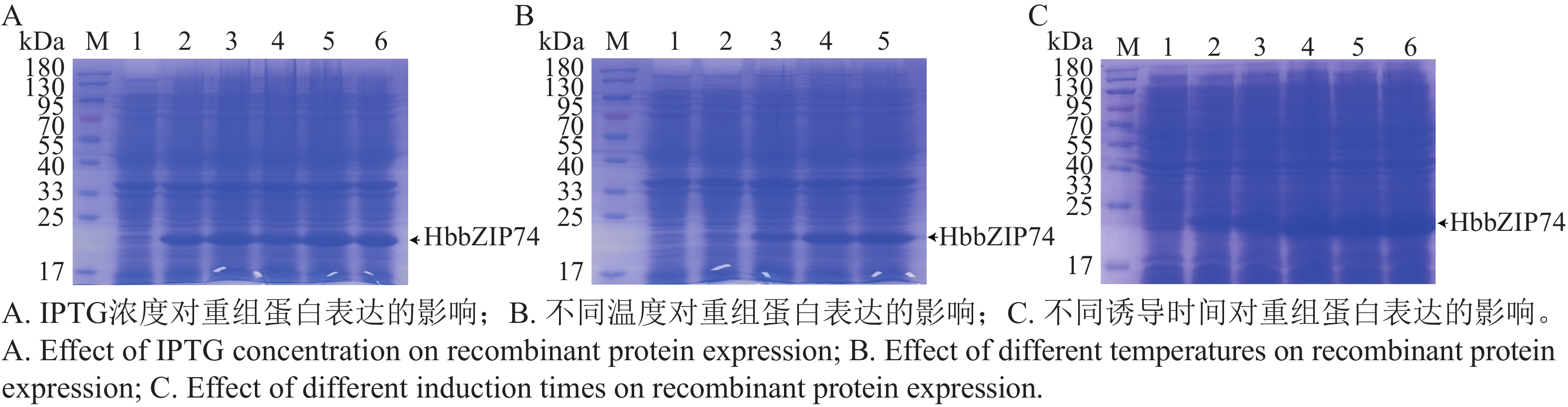
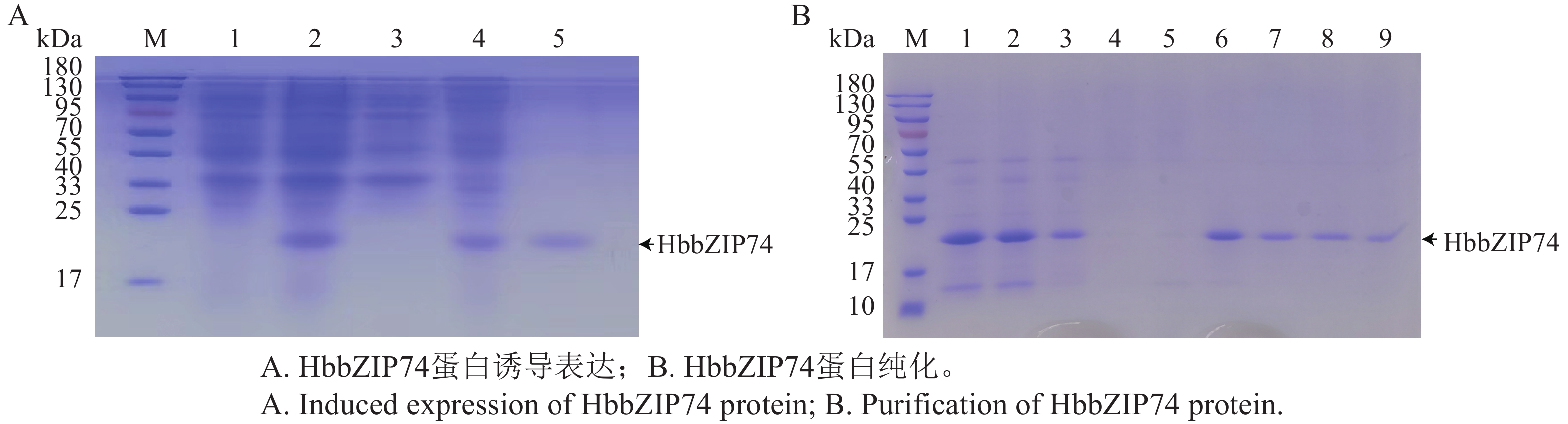
In order to assess the role of the bZIP transcription factor in the biosynthesis of natural rubber in Hevea brasiliensis, HbbZIP74 gene was successfully cloned based on previous work in our laboratory. Bioinformatics analysis and expression pattern analysis were performed. The pET28-HbbZIP74 expression vector was constructed, and the recombinant protein was expressed in the strain Escherichia coli BL21 (DE3) and then purified. The results showed that the open reading frame (ORF) of the gene HbbZIP74 is 486 bp, encoding 161 amino acids, with a bZIP domain, classifying it as a bZIP transcription factor S subgroup. The expression level of HbbZIP74 is higher in latex and leaves, and its expression can be induced by jasmonic acid, abscisic acid, ethylene, and salicylic acid in latex. The optimal condition for the heterologous expression of the HbbZIP74-His recombinant protein was induced at 1 mmol·L−1 IPTG and 37 ℃ for 3 h. The recombinant protein was mainly accumulated in inclusion bodies. The recombinant protein, approximately 22 kDa in size, was purified using Ni-NTA affinity chromatography, which was consistent with expectations. This study lays the foundation for further exploration of the role of HbbZIP74 in the biosynthesis of natural rubber.
In order to assess the role of the bZIP transcription factor in the biosynthesis of natural rubber in Hevea brasiliensis, HbbZIP74 gene was successfully cloned based on previous work in our laboratory. Bioinformatics analysis and expression pattern analysis were performed. The pET28-HbbZIP74 expression vector was constructed, and the recombinant protein was expressed in the strain Escherichia coli BL21 (DE3) and then purified. The results showed that the open reading frame (ORF) of the gene HbbZIP74 is 486 bp, encoding 161 amino acids, with a bZIP domain, classifying it as a bZIP transcription factor S subgroup. The expression level of HbbZIP74 is higher in latex and leaves, and its expression can be induced by jasmonic acid, abscisic acid, ethylene, and salicylic acid in latex. The optimal condition for the heterologous expression of the HbbZIP74-His recombinant protein was induced at 1 mmol·L−1 IPTG and 37 ℃ for 3 h. The recombinant protein was mainly accumulated in inclusion bodies. The recombinant protein, approximately 22 kDa in size, was purified using Ni-NTA affinity chromatography, which was consistent with expectations. This study lays the foundation for further exploration of the role of HbbZIP74 in the biosynthesis of natural rubber.

2026, 17(3): 410-421.
doi: 10.15886/j.cnki.rdswxb.20250184
Abstract:
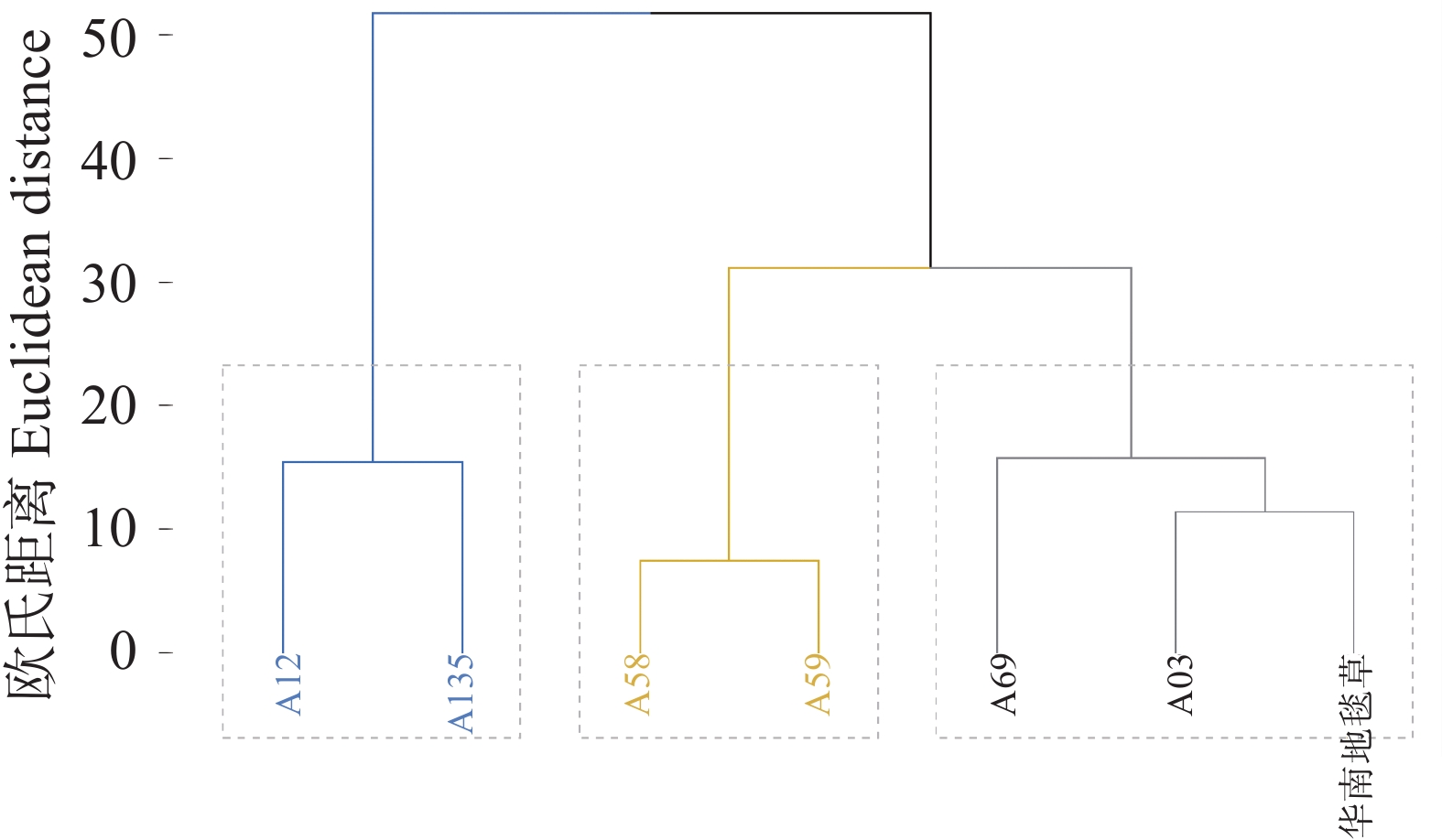
To investigate the shade tolerance of Axonopus compressus, shade tolerance evaluation was performed on six accessions of superior Axonopus compressus germplasm (A03, A12, A58, A59, A69, A135) with Axonopus compressus ‘Huanan’ as the control variety, so as to provide a theoretical basis for breeding shade-tolerant varieties of A. compressus. A. compressus was treated with 75% shading or natural light under hydroponic culture and 18 indicators including chlorophyll content, leaf length, leaf width, biomass, and turf quality were measured after 28 days of treatment for comprehensive evaluation. The results show that compared to the control group, the 75% shading stress treatment, significantly increased the leaf length, but significantly decreased, leaf width, chlorophyll content, turf height, biomass, and turf quality of A. compressus. Comprehensive analysis showed that the germplasm accessions were classified into three categories: shade-tolerant, moderately shade-tolerant, and shade-sensitive. Among them, Accession A69 exhibited the highest shade tolerance and was listed in, the shade-tolerant category along with Accession A03 and the control variety; Accessions A58 and A59 were listed in the moderately shade-tolerant category; Accessions A12 and A135 were listed in the shade-sensitive category, demonstrating the poorest shade tolerance.
To investigate the shade tolerance of Axonopus compressus, shade tolerance evaluation was performed on six accessions of superior Axonopus compressus germplasm (A03, A12, A58, A59, A69, A135) with Axonopus compressus ‘Huanan’ as the control variety, so as to provide a theoretical basis for breeding shade-tolerant varieties of A. compressus. A. compressus was treated with 75% shading or natural light under hydroponic culture and 18 indicators including chlorophyll content, leaf length, leaf width, biomass, and turf quality were measured after 28 days of treatment for comprehensive evaluation. The results show that compared to the control group, the 75% shading stress treatment, significantly increased the leaf length, but significantly decreased, leaf width, chlorophyll content, turf height, biomass, and turf quality of A. compressus. Comprehensive analysis showed that the germplasm accessions were classified into three categories: shade-tolerant, moderately shade-tolerant, and shade-sensitive. Among them, Accession A69 exhibited the highest shade tolerance and was listed in, the shade-tolerant category along with Accession A03 and the control variety; Accessions A58 and A59 were listed in the moderately shade-tolerant category; Accessions A12 and A135 were listed in the shade-sensitive category, demonstrating the poorest shade tolerance.


2026, 17(3): 422-430.
doi: 10.15886/j.cnki.rdswxb.20250126
Abstract:
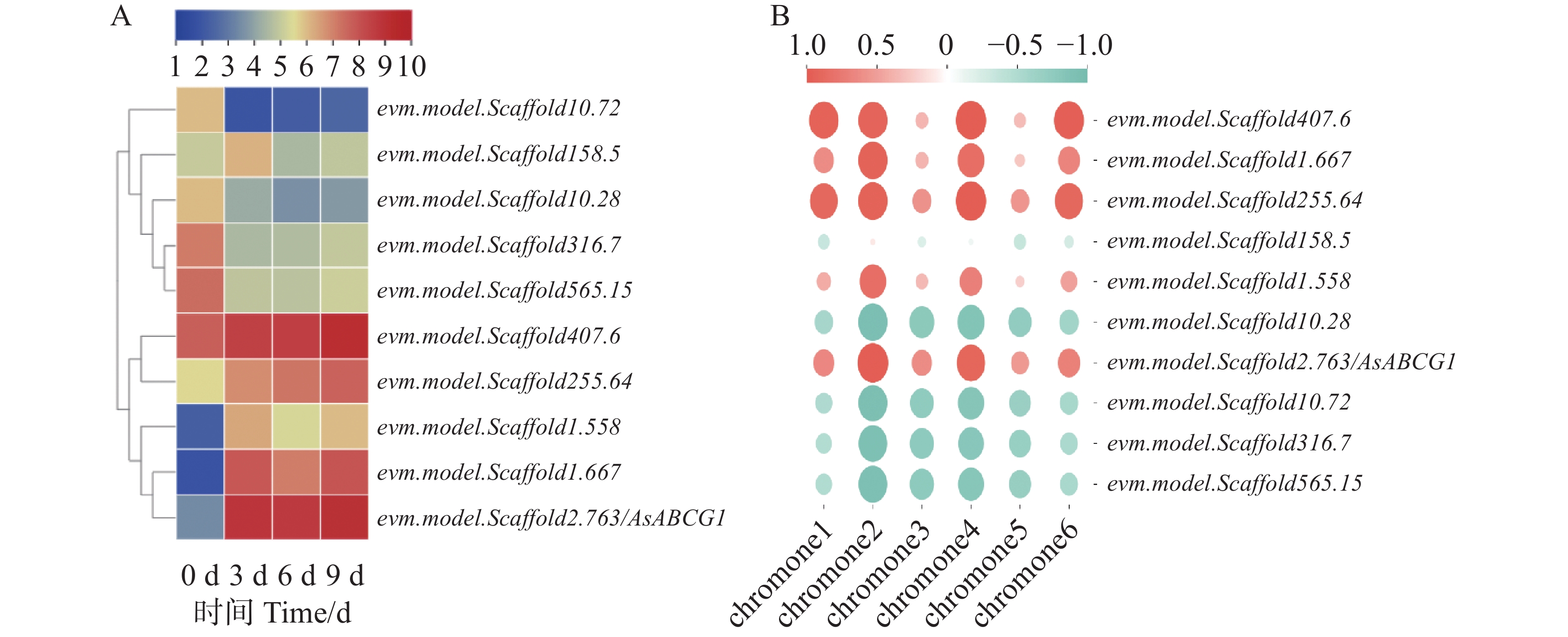
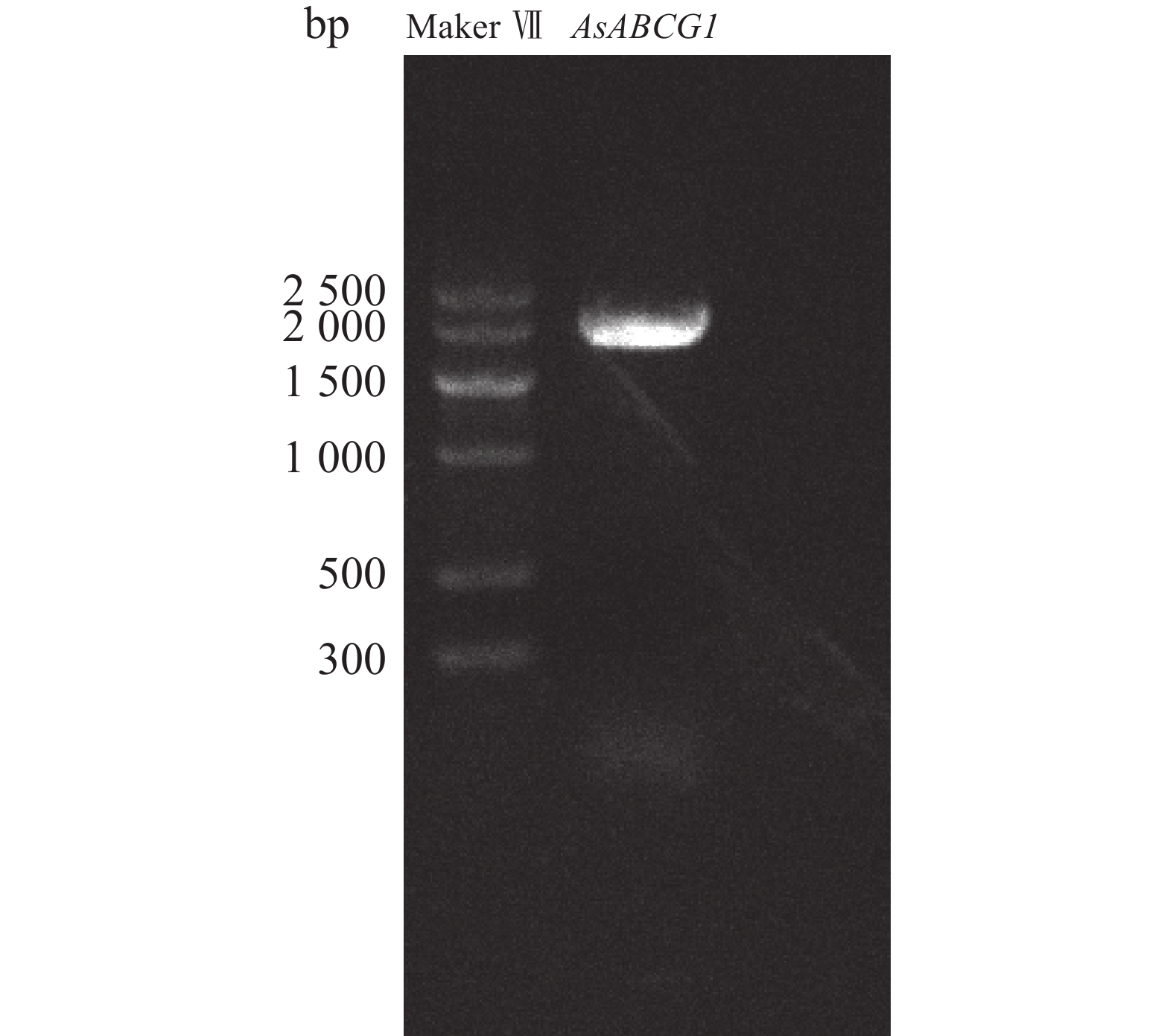
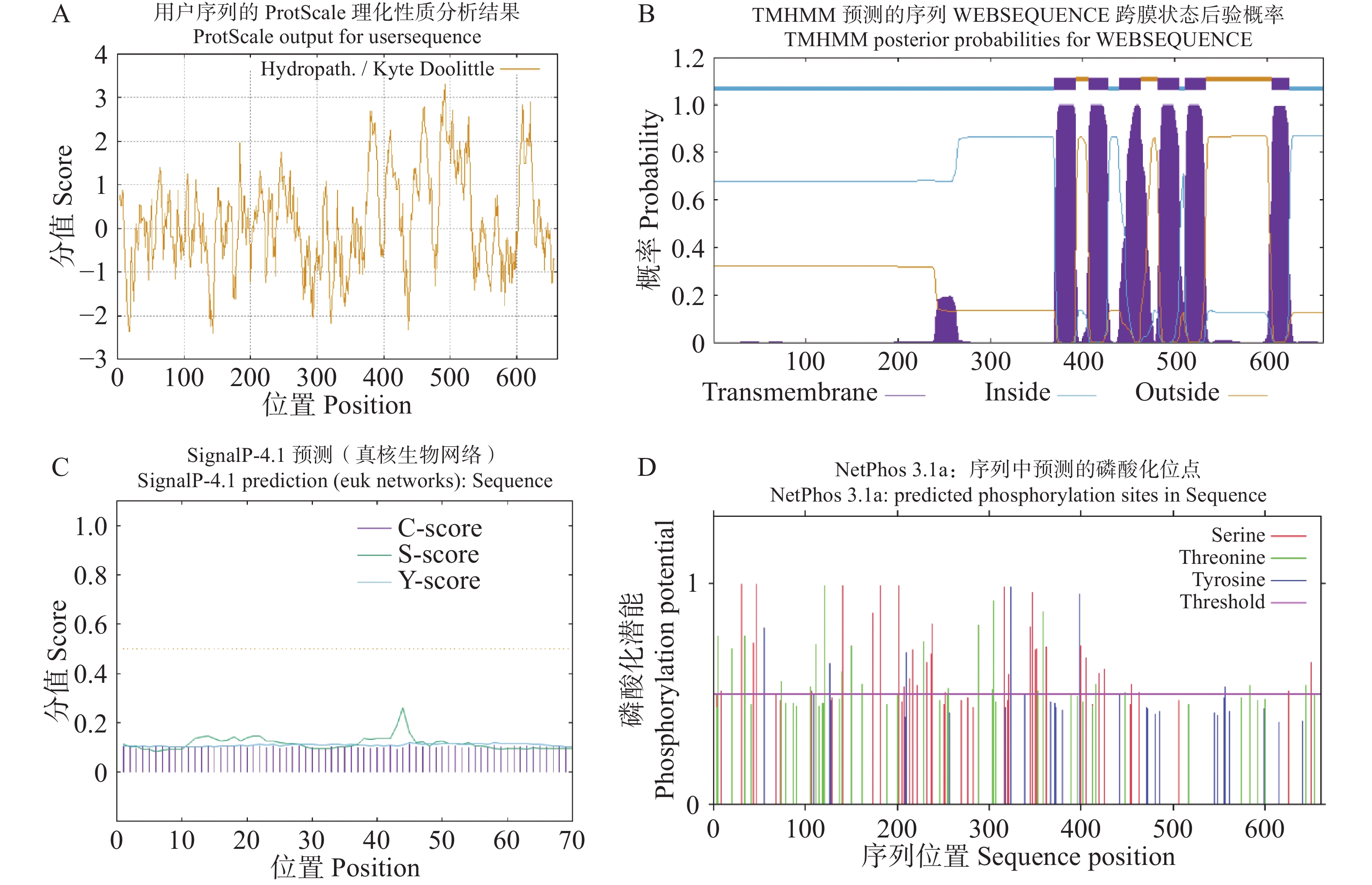
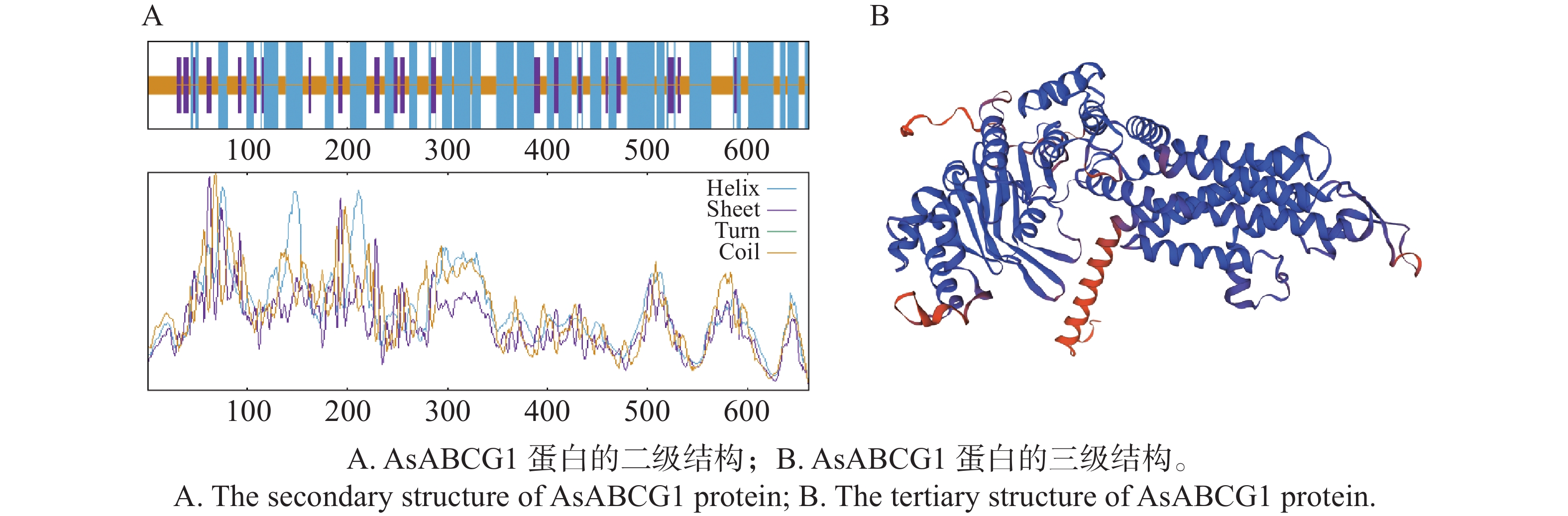
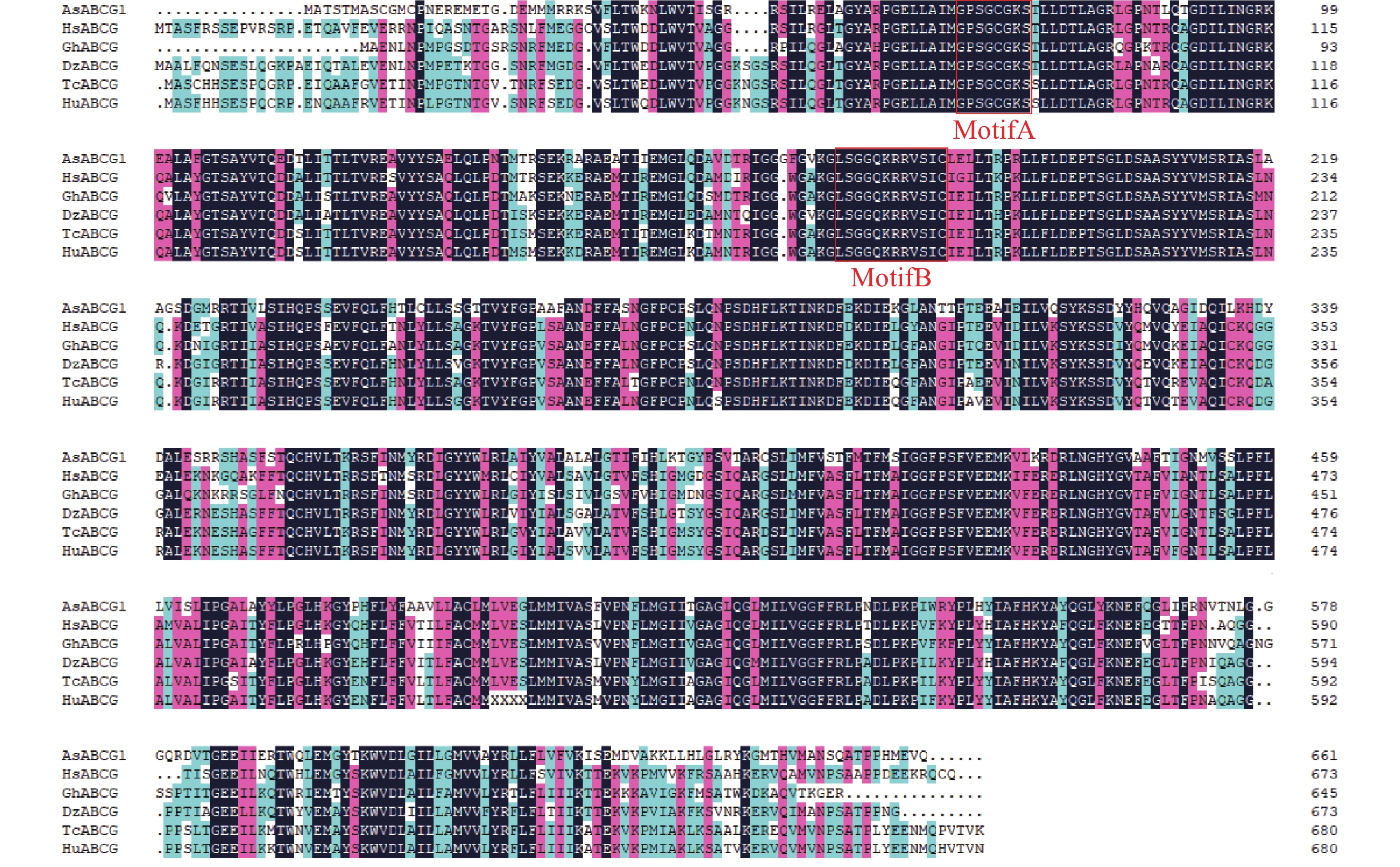
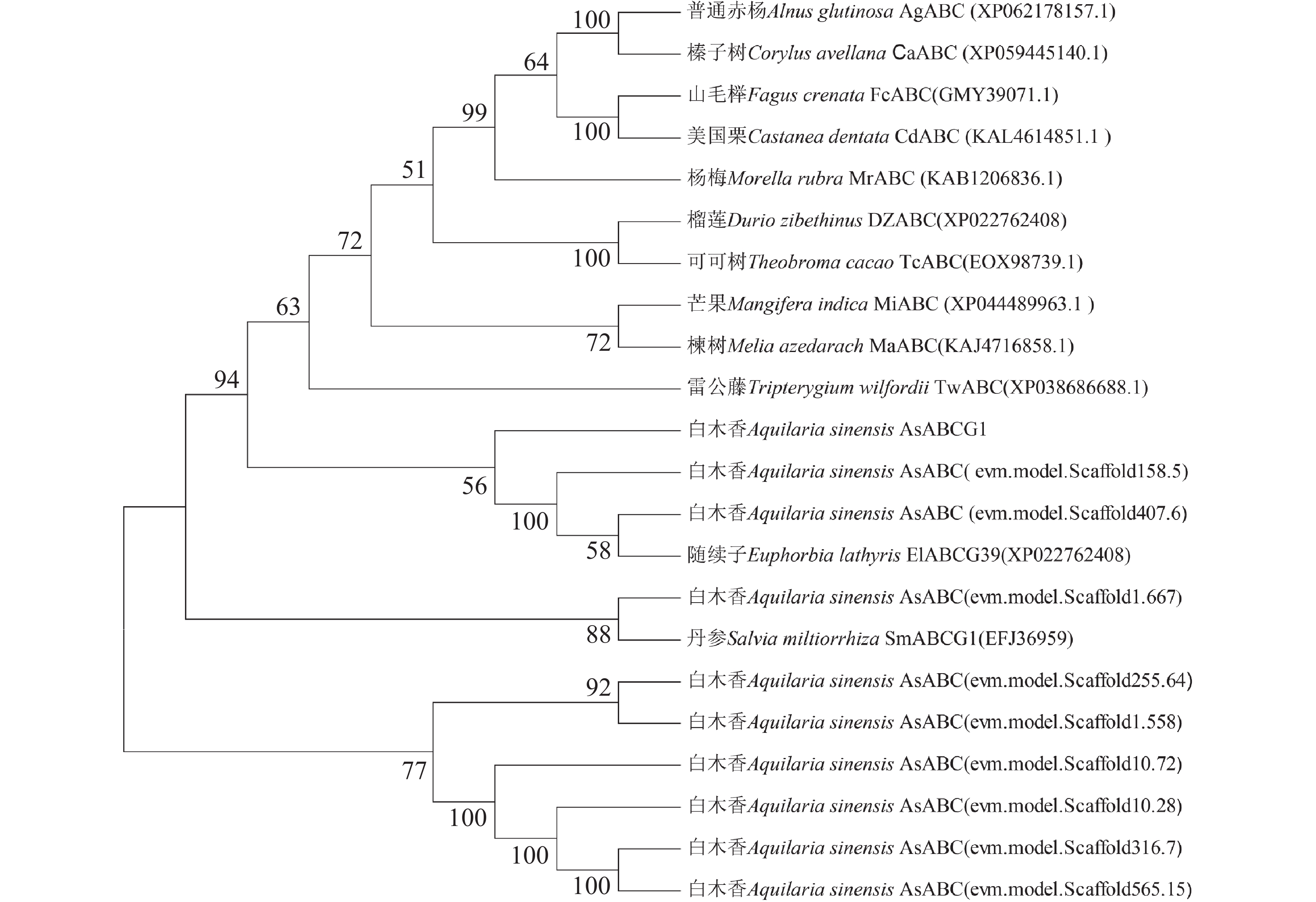
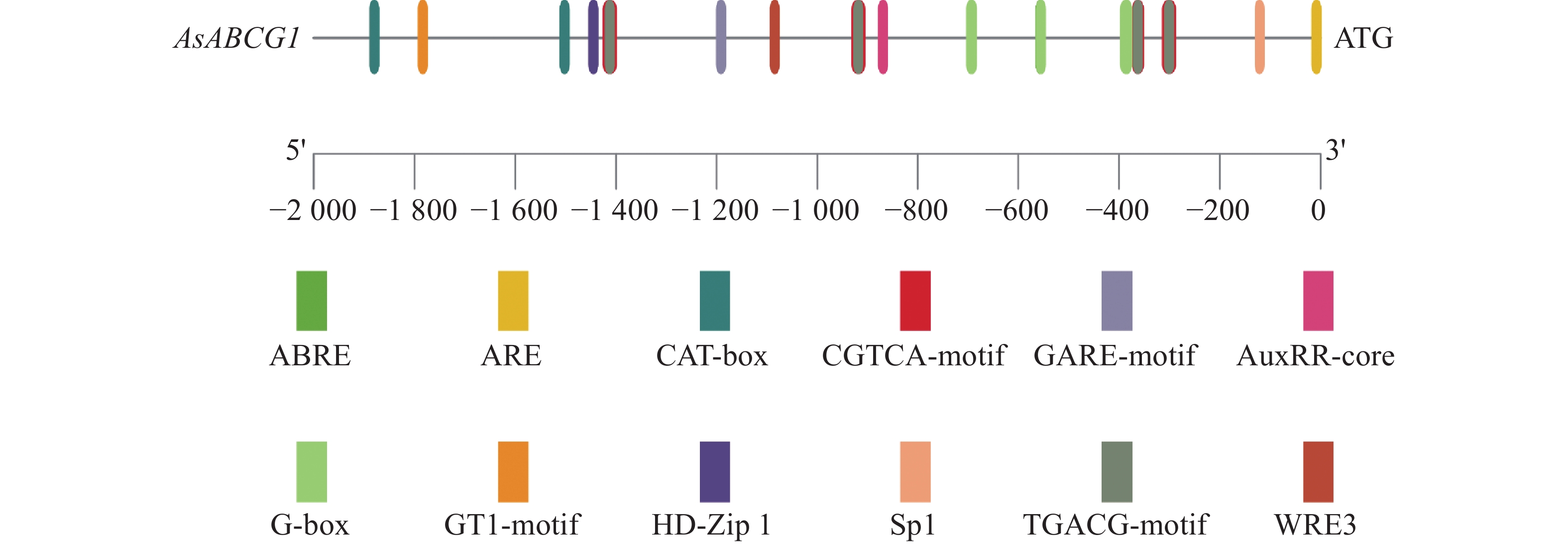
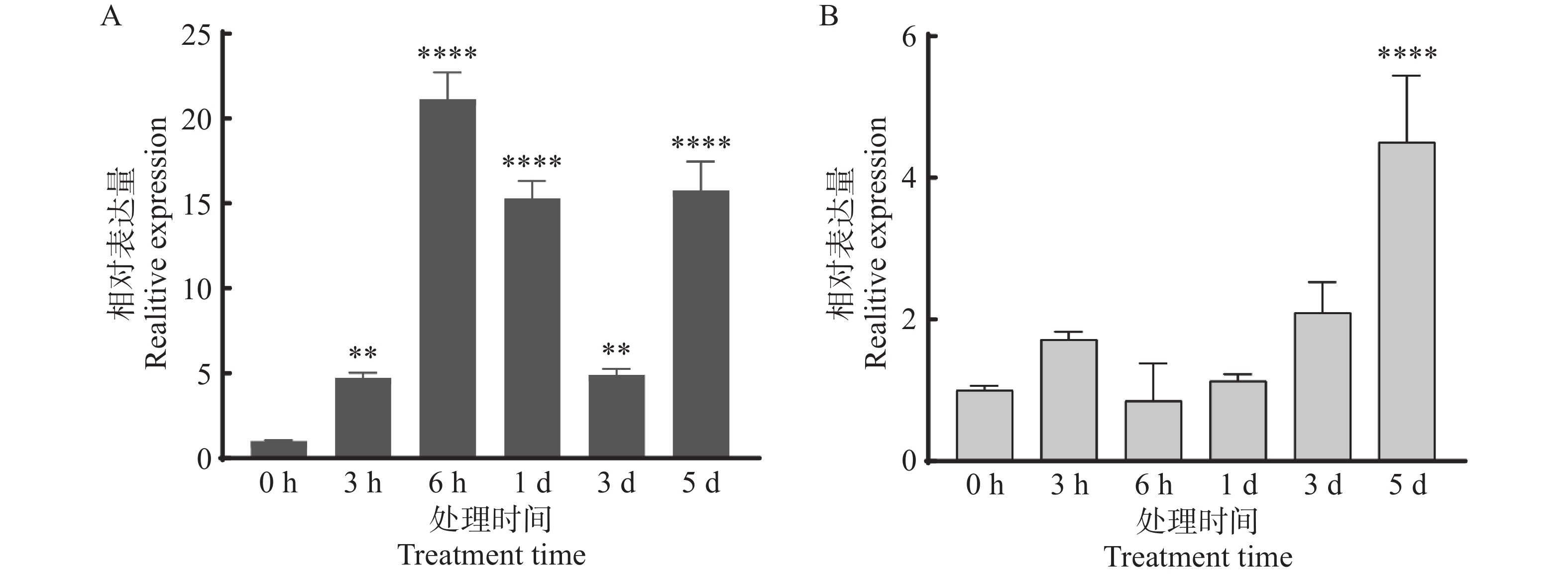
Agarwood is a traditional precious medicinal material and a natural aromatic material, which is highly valuable due to its rich secondary metabolites. ABC transporters are a type of transmembrane proteins widely distributed in organisms and play a crucial role in the transport and accumulation of secondary metabolites in plants. To deeply explore the function of ABC transporters in the transport process of secondary metabolites in agarwood, a gene encoding an ABC transporter AsABCG1; was identified by analyzing the transcriptome data of Aquilaria sinensis. The coding sequence (CDS) length of this gene is 1986 bp, encoding a protein consisting of 661 amino acids. AsABCG1 has typical structural characteristics of plant ABC transporters, including two conserved domains, Motif A and Motif B. The promoter region of AsABCG1 contains various hormone response elements such as jasmonic acid, auxin, gibberellin, abscisic acid, as well as stress response elements such as anaerobic induction and mechanical injury. The real-time fluorescence quantitative PCR (RT-qPCR) analysis showed that methyl jasmonic acid and mechanical injury treatments could significantly induce the expression of AsABCG1 in the stems of A. sinensis. All these findings laid the foundation for further elucidating the function of the AsABCG1 gene in the defense response to stress and the accumulation of secondary metabolites in A. sinensis.
Agarwood is a traditional precious medicinal material and a natural aromatic material, which is highly valuable due to its rich secondary metabolites. ABC transporters are a type of transmembrane proteins widely distributed in organisms and play a crucial role in the transport and accumulation of secondary metabolites in plants. To deeply explore the function of ABC transporters in the transport process of secondary metabolites in agarwood, a gene encoding an ABC transporter AsABCG1; was identified by analyzing the transcriptome data of Aquilaria sinensis. The coding sequence (CDS) length of this gene is 1986 bp, encoding a protein consisting of 661 amino acids. AsABCG1 has typical structural characteristics of plant ABC transporters, including two conserved domains, Motif A and Motif B. The promoter region of AsABCG1 contains various hormone response elements such as jasmonic acid, auxin, gibberellin, abscisic acid, as well as stress response elements such as anaerobic induction and mechanical injury. The real-time fluorescence quantitative PCR (RT-qPCR) analysis showed that methyl jasmonic acid and mechanical injury treatments could significantly induce the expression of AsABCG1 in the stems of A. sinensis. All these findings laid the foundation for further elucidating the function of the AsABCG1 gene in the defense response to stress and the accumulation of secondary metabolites in A. sinensis.
2026, 17(3): 431-438.
doi: 10.15886/j.cnki.rdswxb.20250106
Abstract:
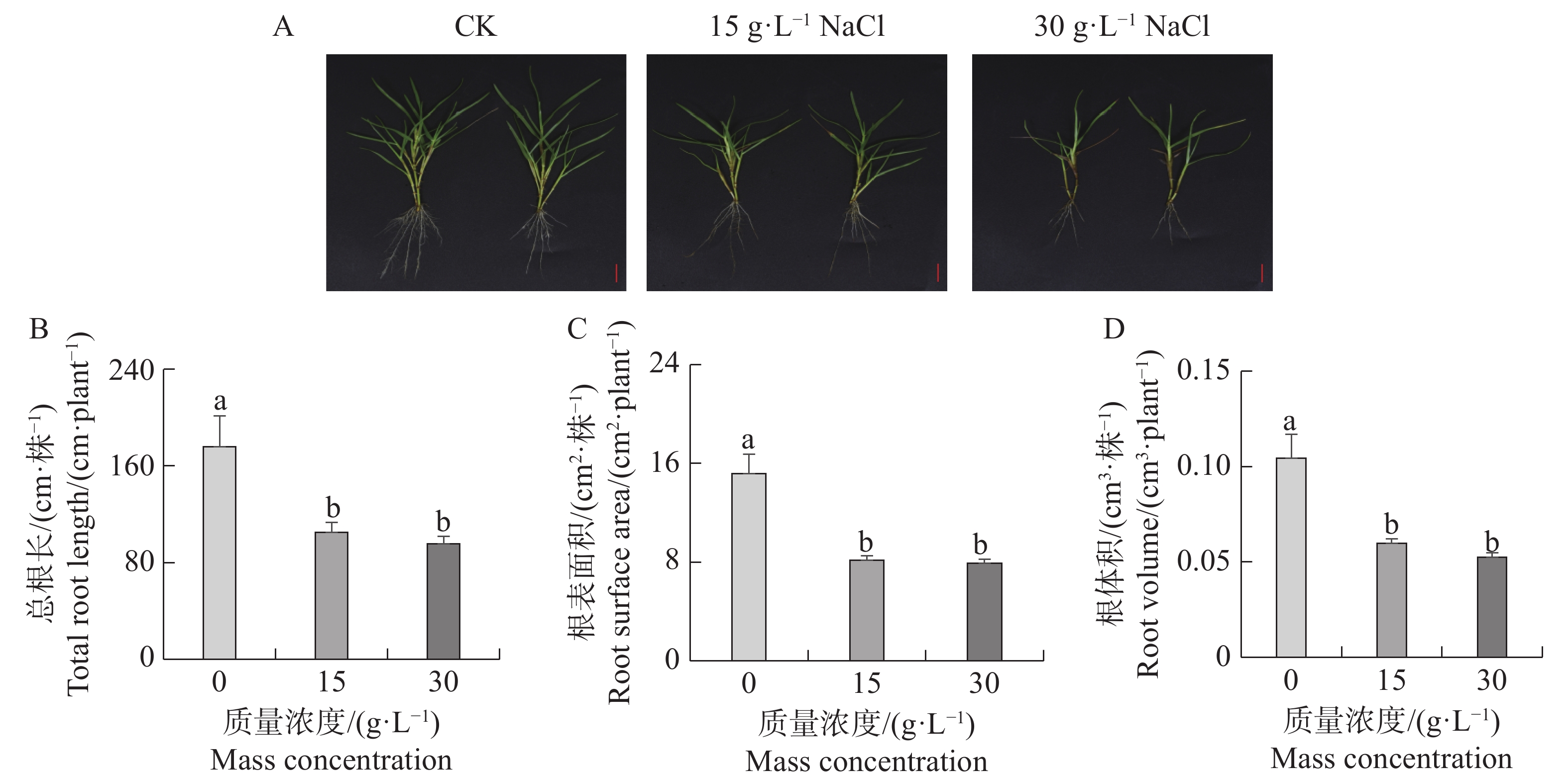
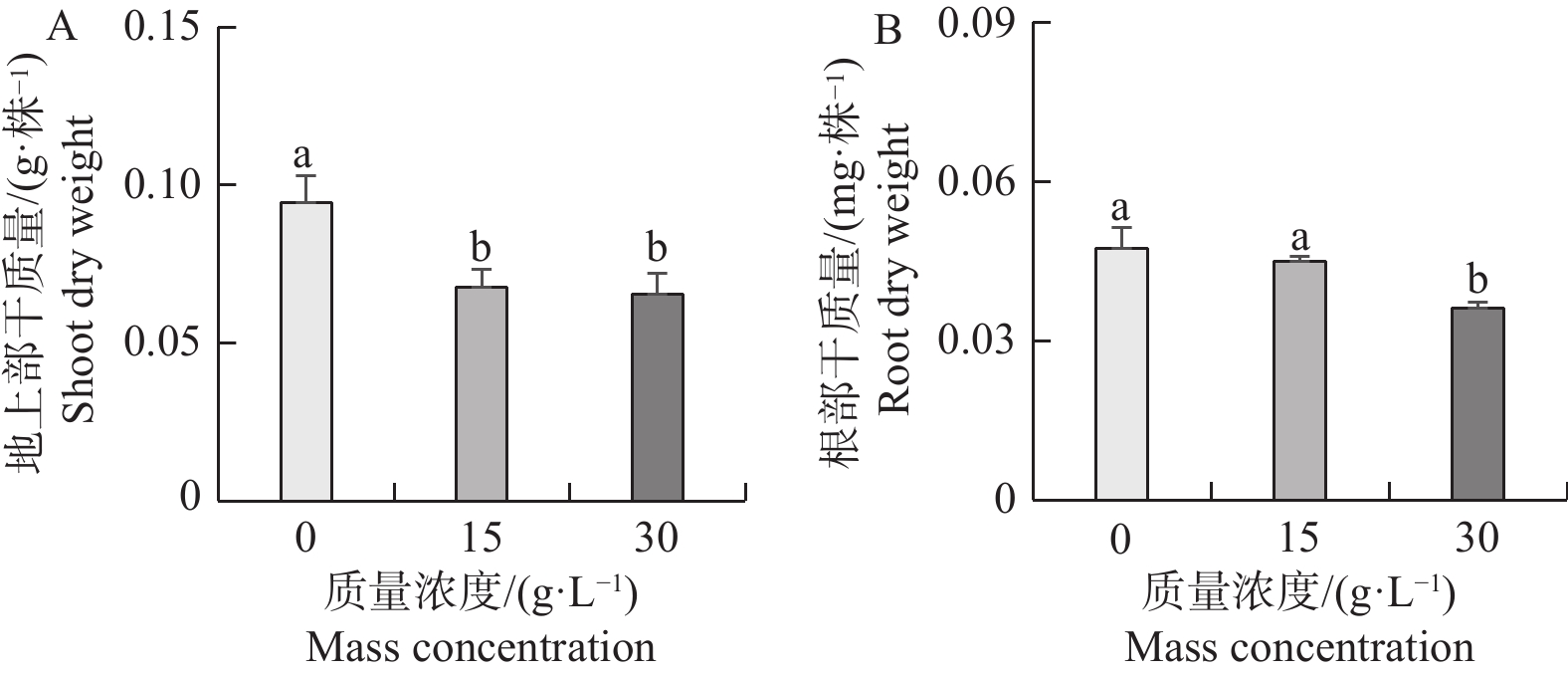
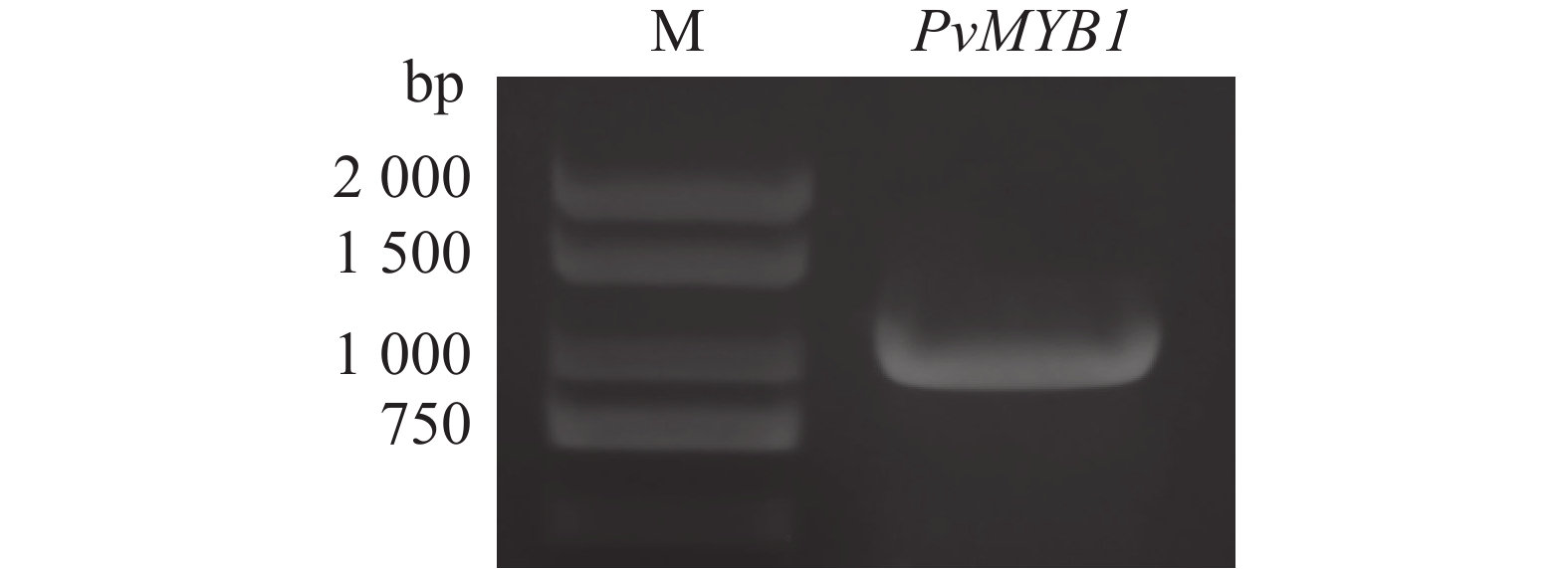
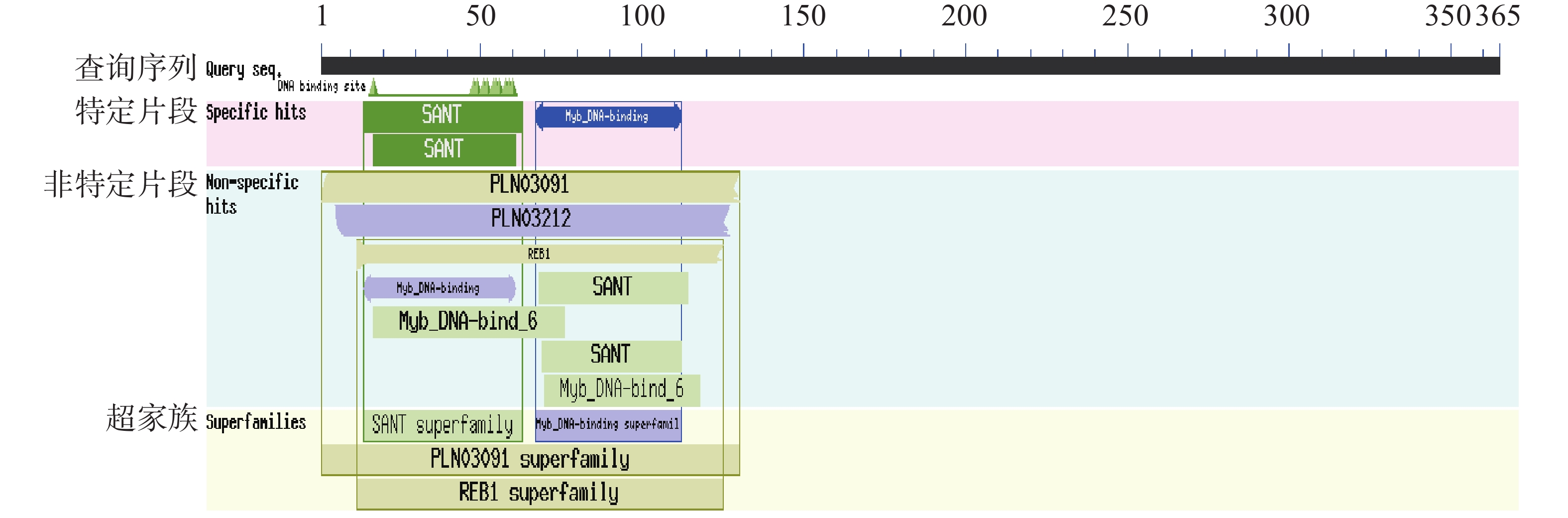
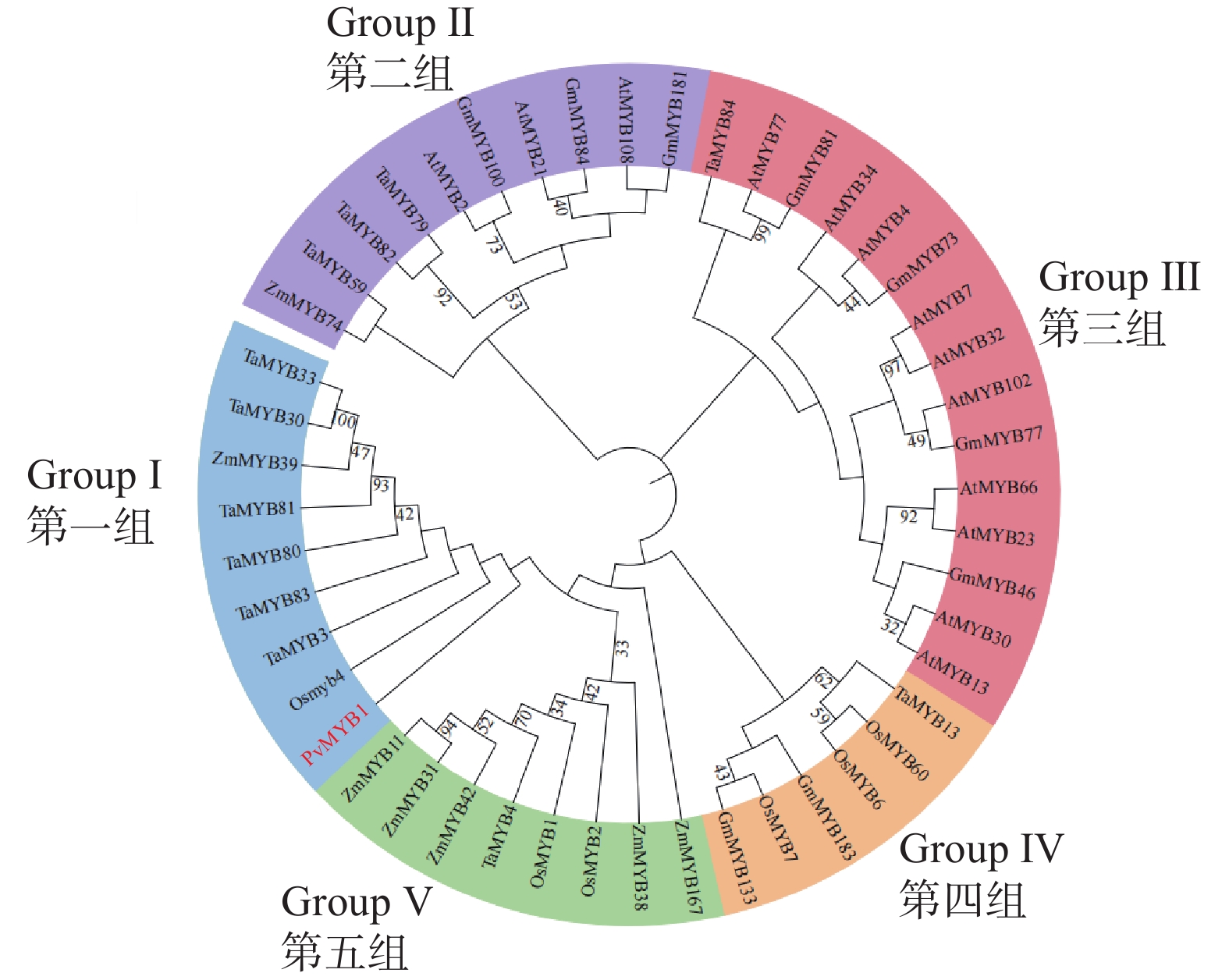
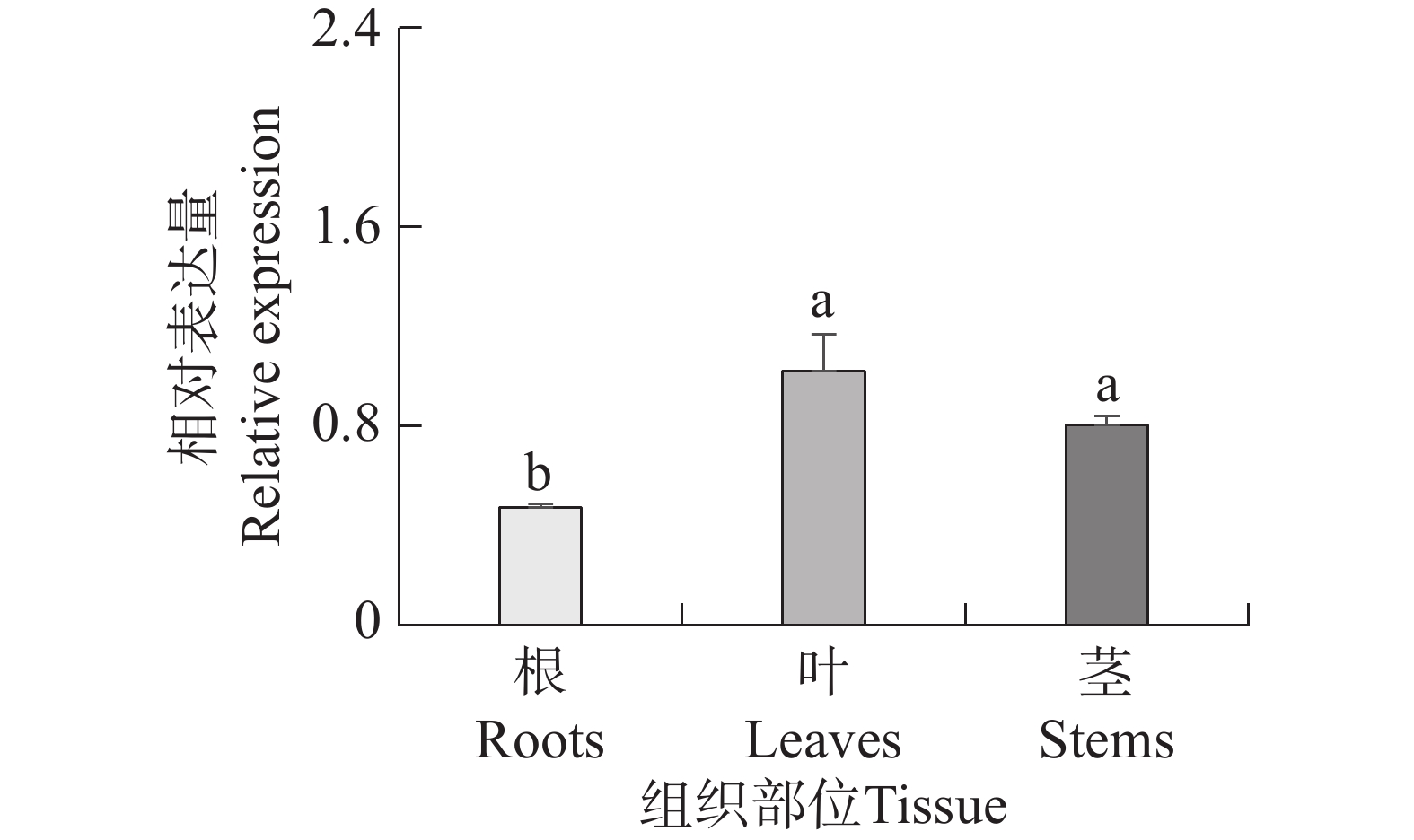
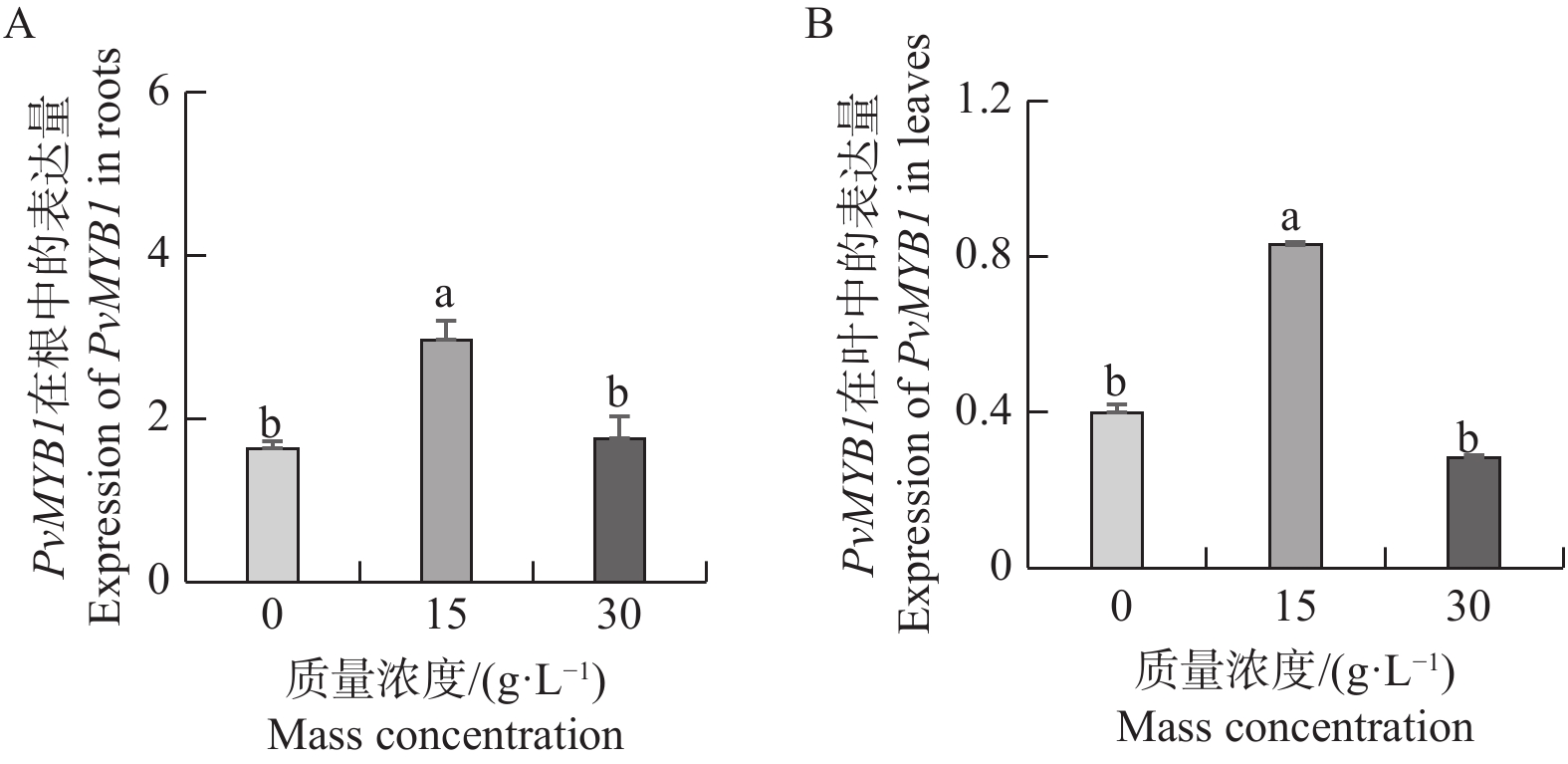
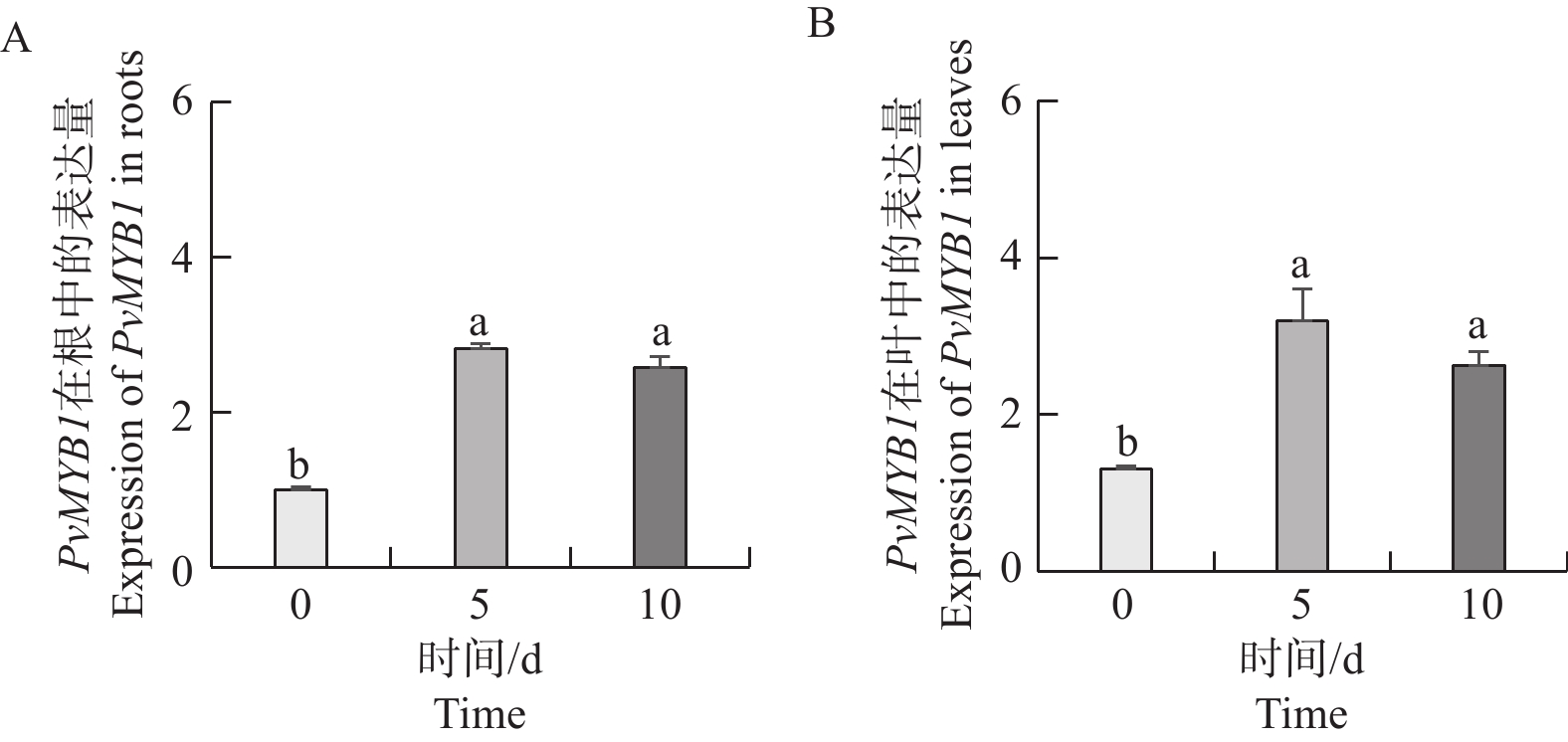
Seashore paspalum (Paspalum vaginatum Sw.) were treated with different concentrations of salt to observe the effect of salt treatment on the growth of seashore paspalum, and the PvMYB1 gene was cloned from seashore paspalum under different treatments and analyzed by using bioinformatics and gene expression analysis methods in a view to investigating whether MYB transcription factor genes are involved in the regulation of growth and development of seashore paspalum under salt stress. The results showed that salt stress inhibited the growth of both shoots and roots in seashore paspalum, manifested by reduction in total root length, root surface area, and root volume. The full length of the seashore paspalum PvMYB1 gene was1098 bp, encoding 365 amino acid residues, with a protein molecular weight of 39.26 kDa, and.this gene belongs to the R2R3-MYB family. Subcellular localization prediction indicated that the PvMYB1 protein was located in the nucleus. RT-qPCR analysis demonstrated that the expression of PvMYB1 was higher in leaves than in roots. The results of salt treatment at different time periods showed that 30 g·L-1 salt treatment significantly increased the transcription level of the PvMYB1 gene. Moreover, under different salt concentration treatments, the expression levels of the PvMYB1 gene in the roots and leaves were significantly upregulated when the salt concentration was 15 g·L-1. All these results revealed that PvMYB1 is implicated in the response of seashore paspalum to salt stress.
Seashore paspalum (Paspalum vaginatum Sw.) were treated with different concentrations of salt to observe the effect of salt treatment on the growth of seashore paspalum, and the PvMYB1 gene was cloned from seashore paspalum under different treatments and analyzed by using bioinformatics and gene expression analysis methods in a view to investigating whether MYB transcription factor genes are involved in the regulation of growth and development of seashore paspalum under salt stress. The results showed that salt stress inhibited the growth of both shoots and roots in seashore paspalum, manifested by reduction in total root length, root surface area, and root volume. The full length of the seashore paspalum PvMYB1 gene was
2026, 17(3): 439-450.
doi: 10.15886/j.cnki.rdswxb.20250098
Abstract:
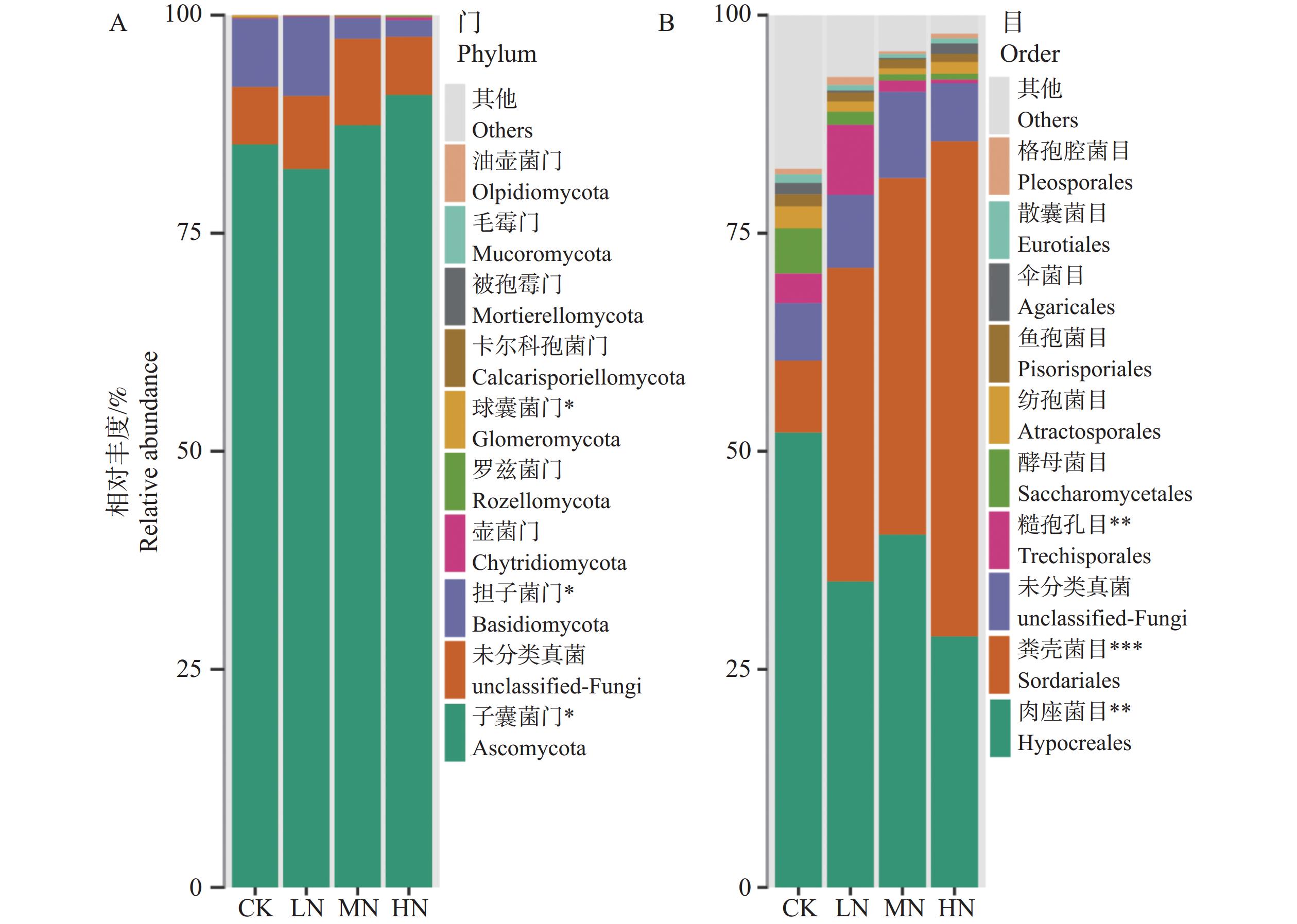
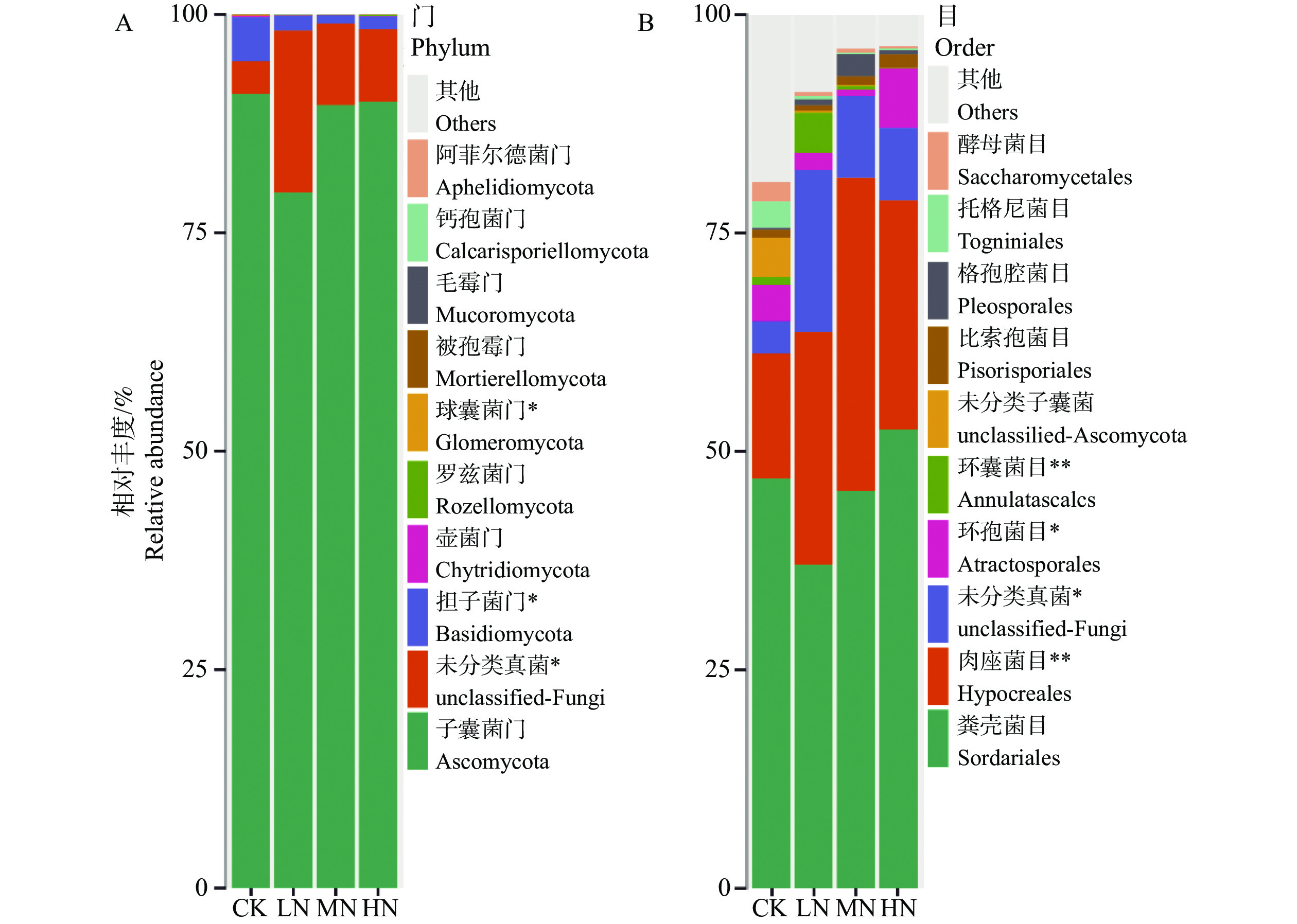
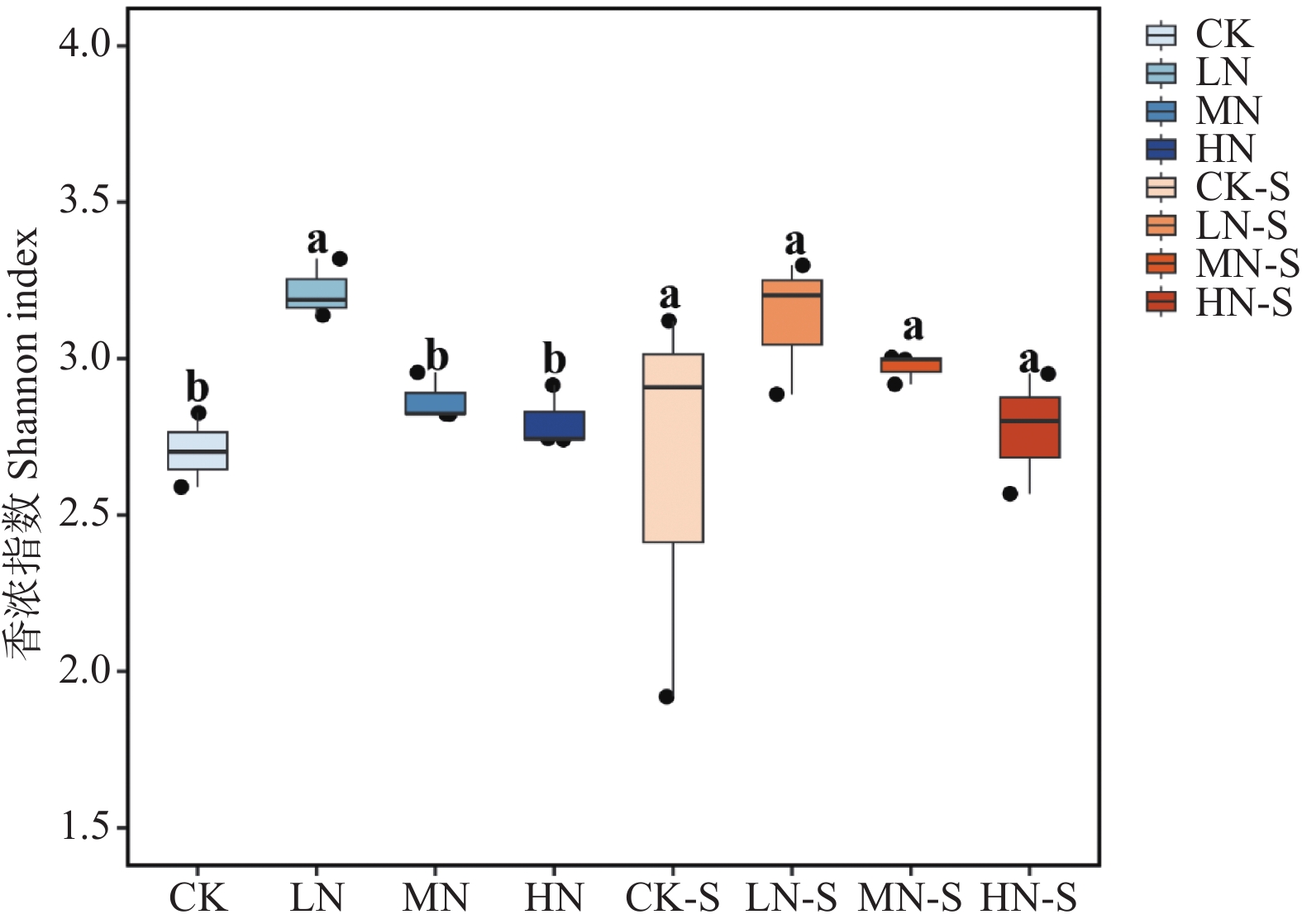
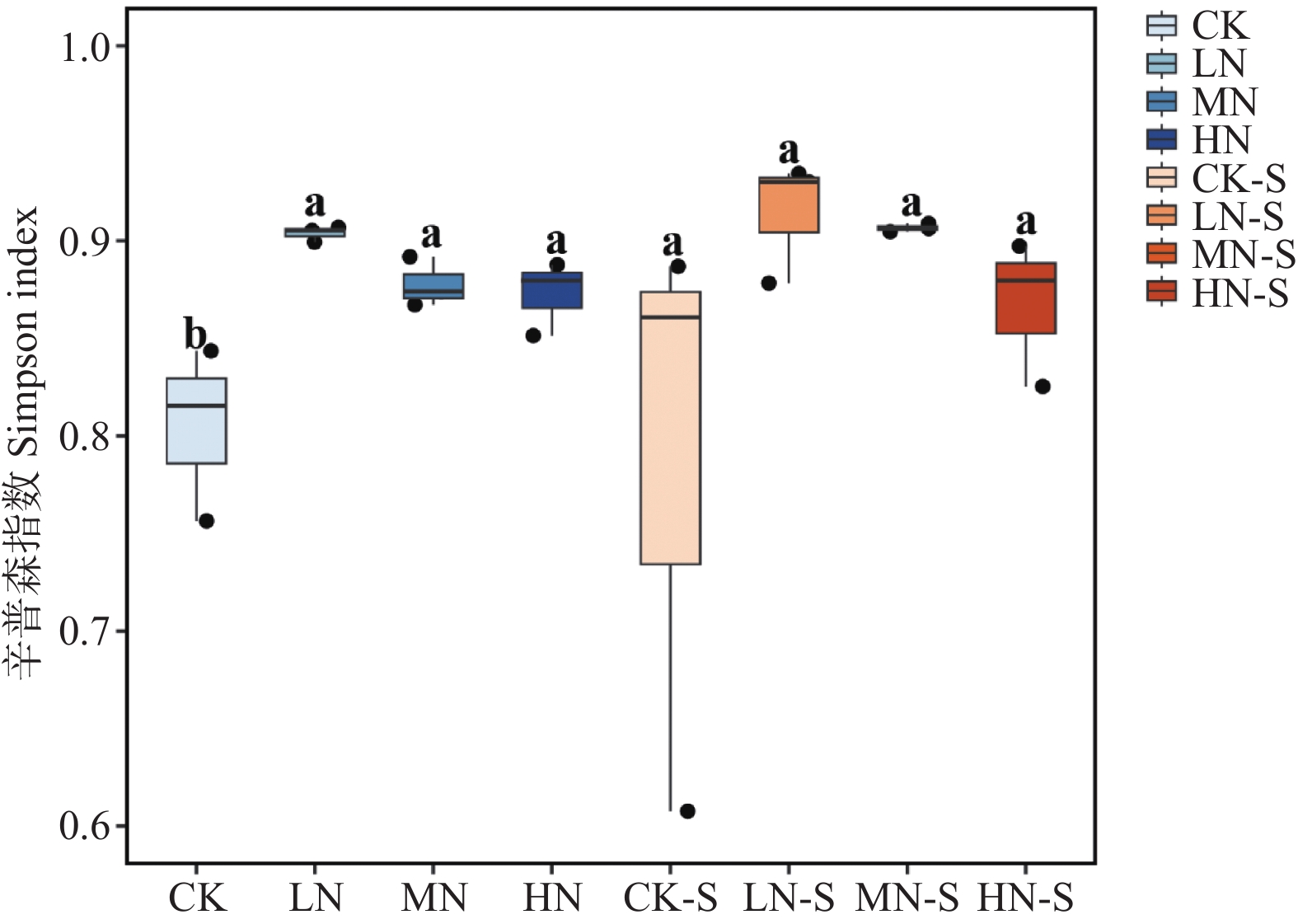
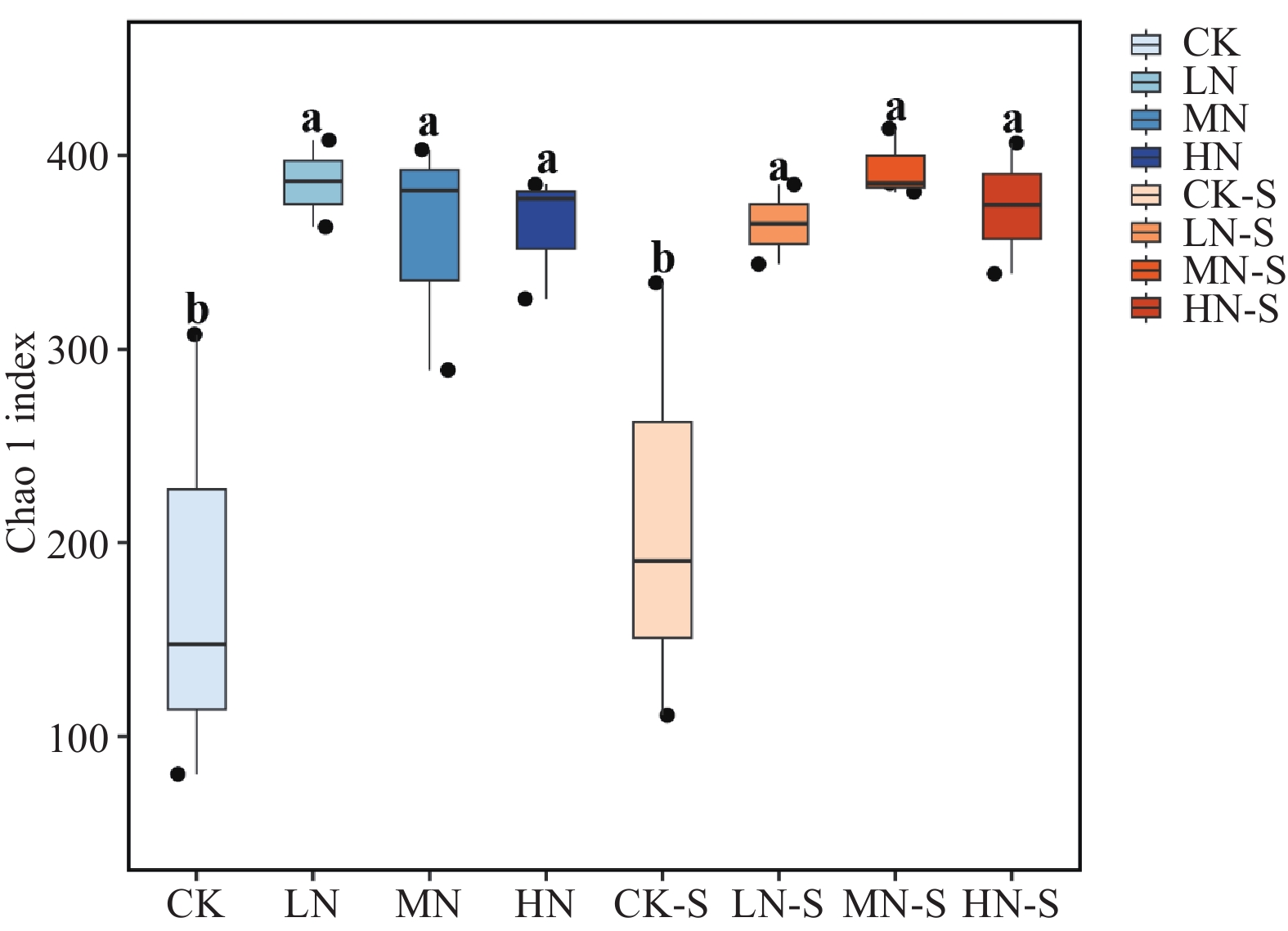
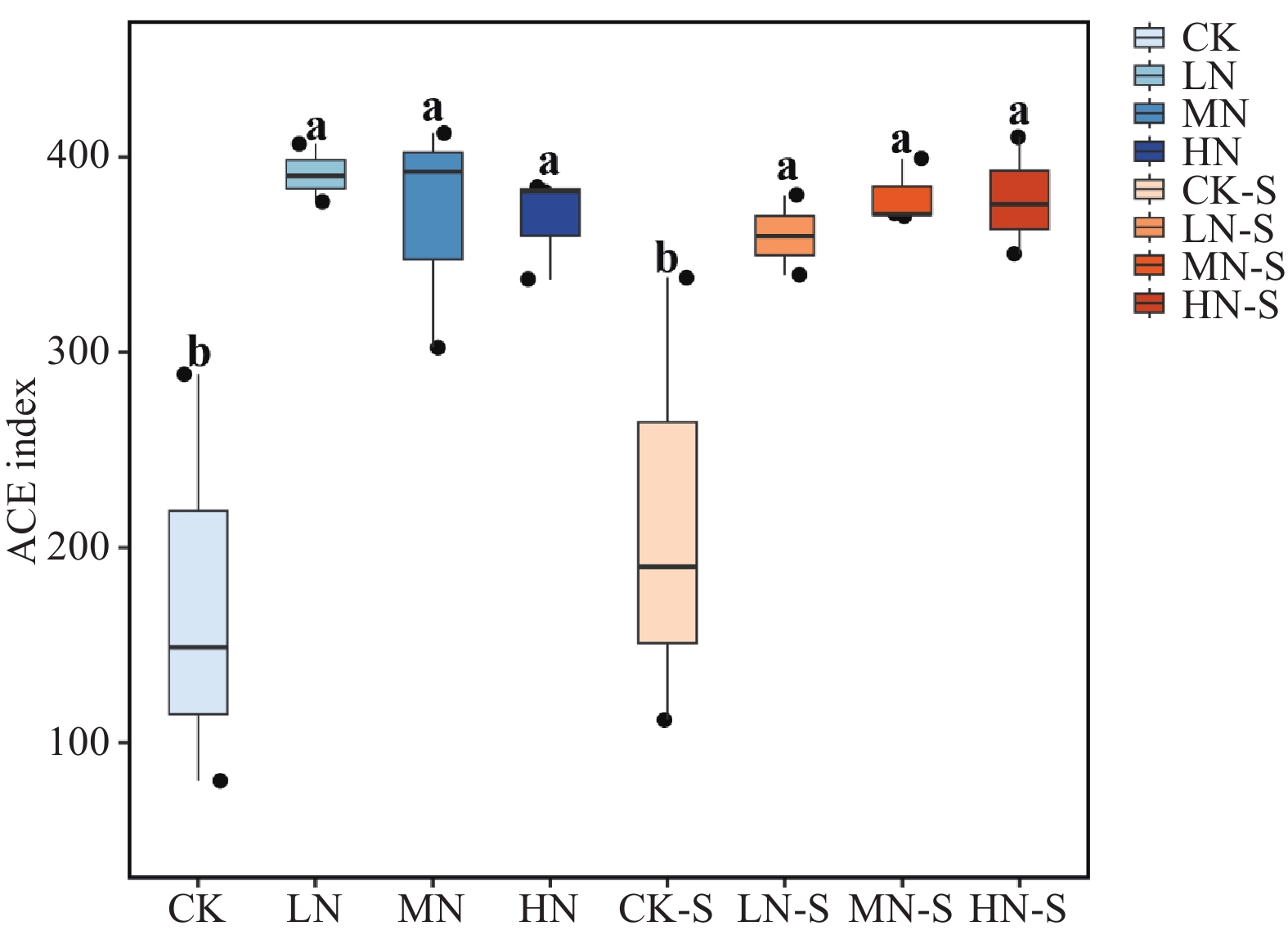
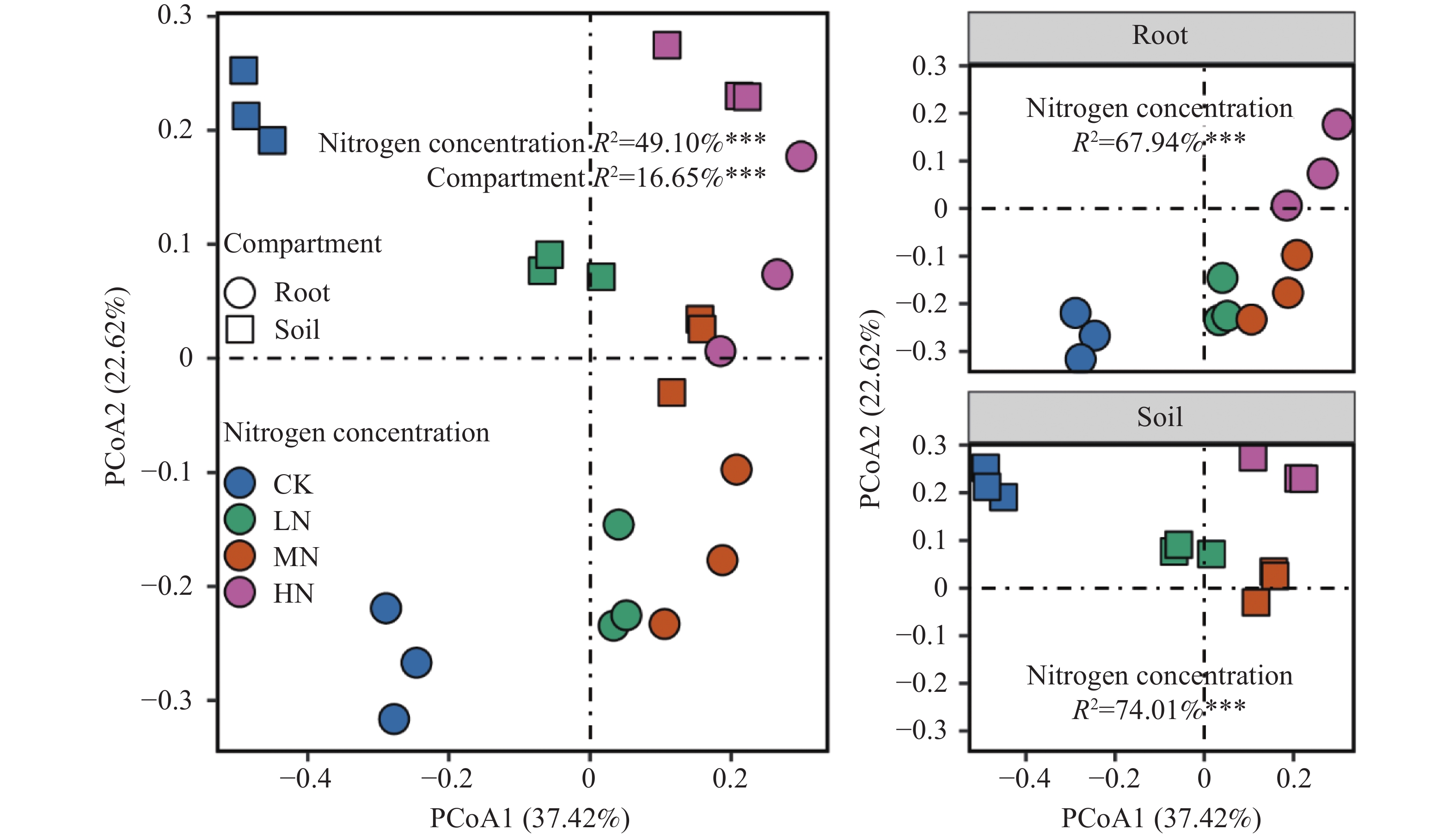
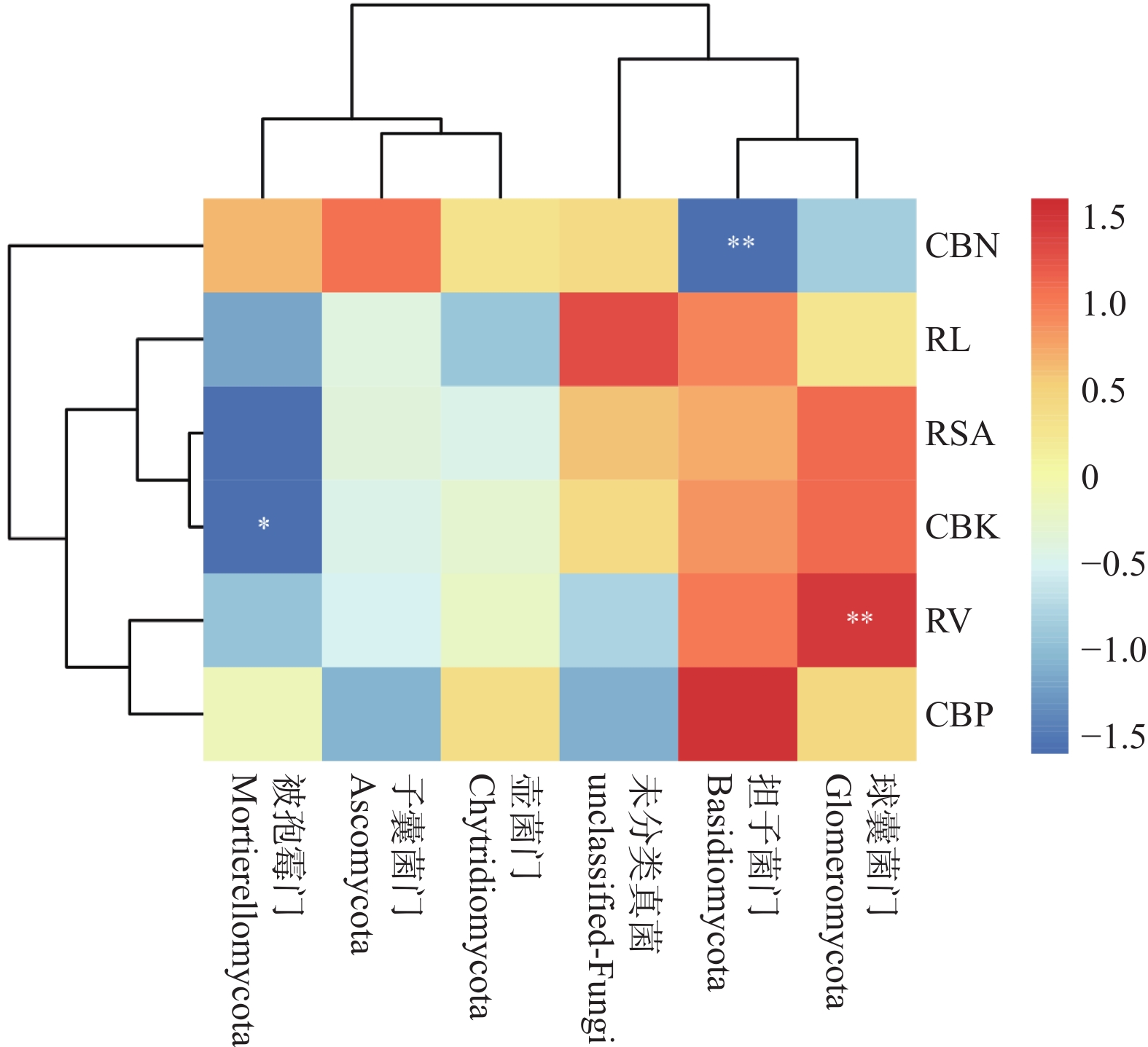
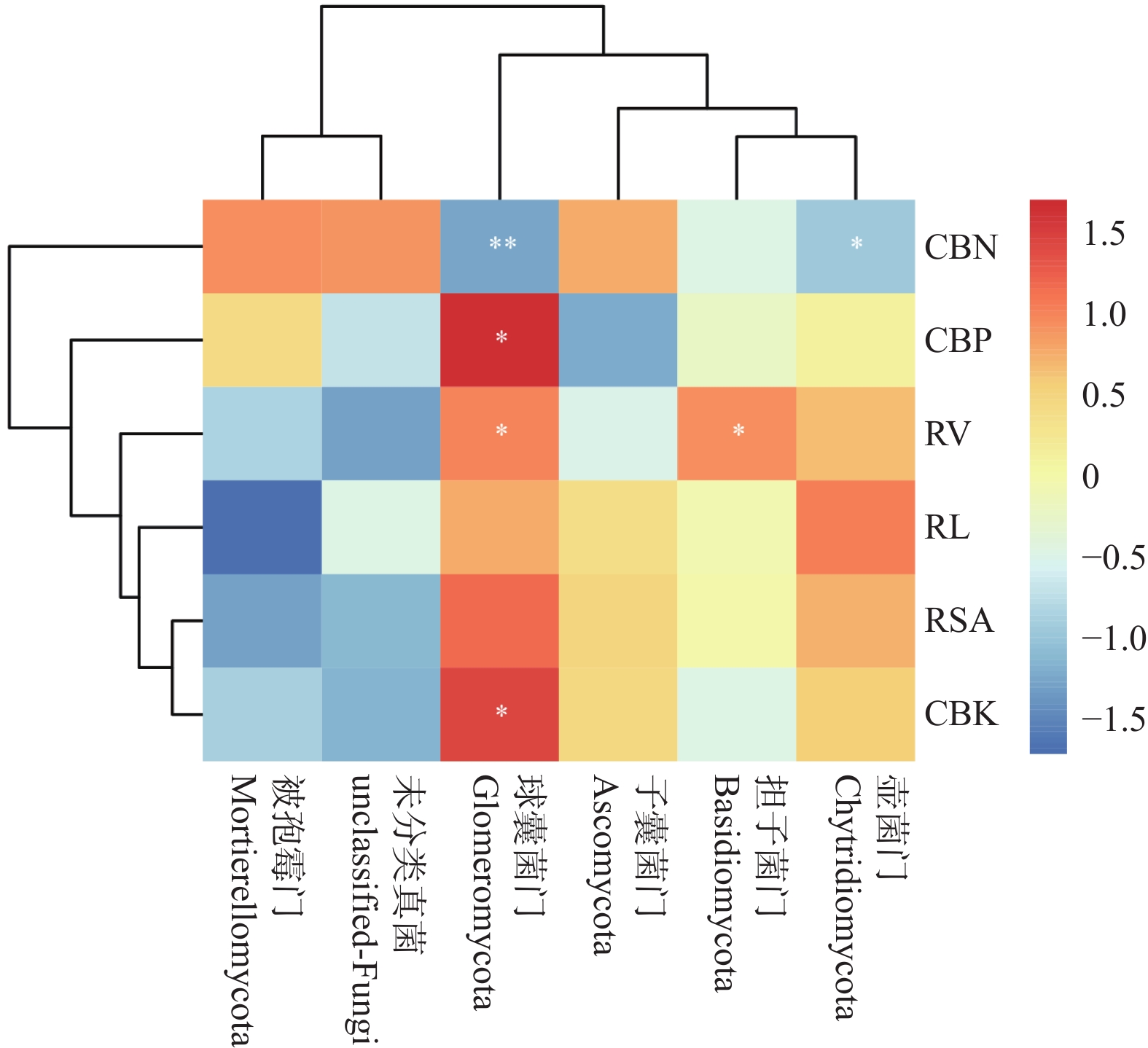
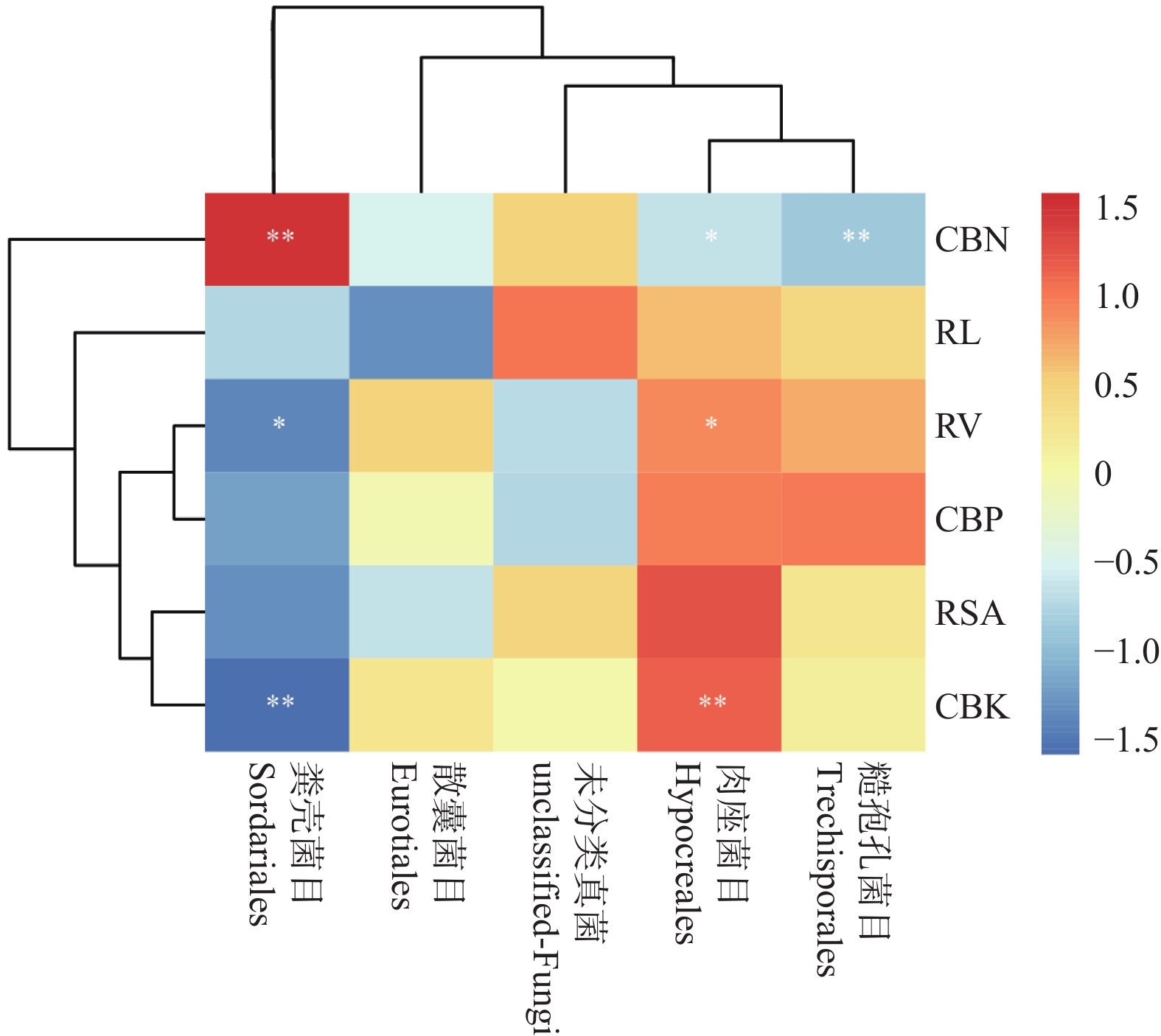
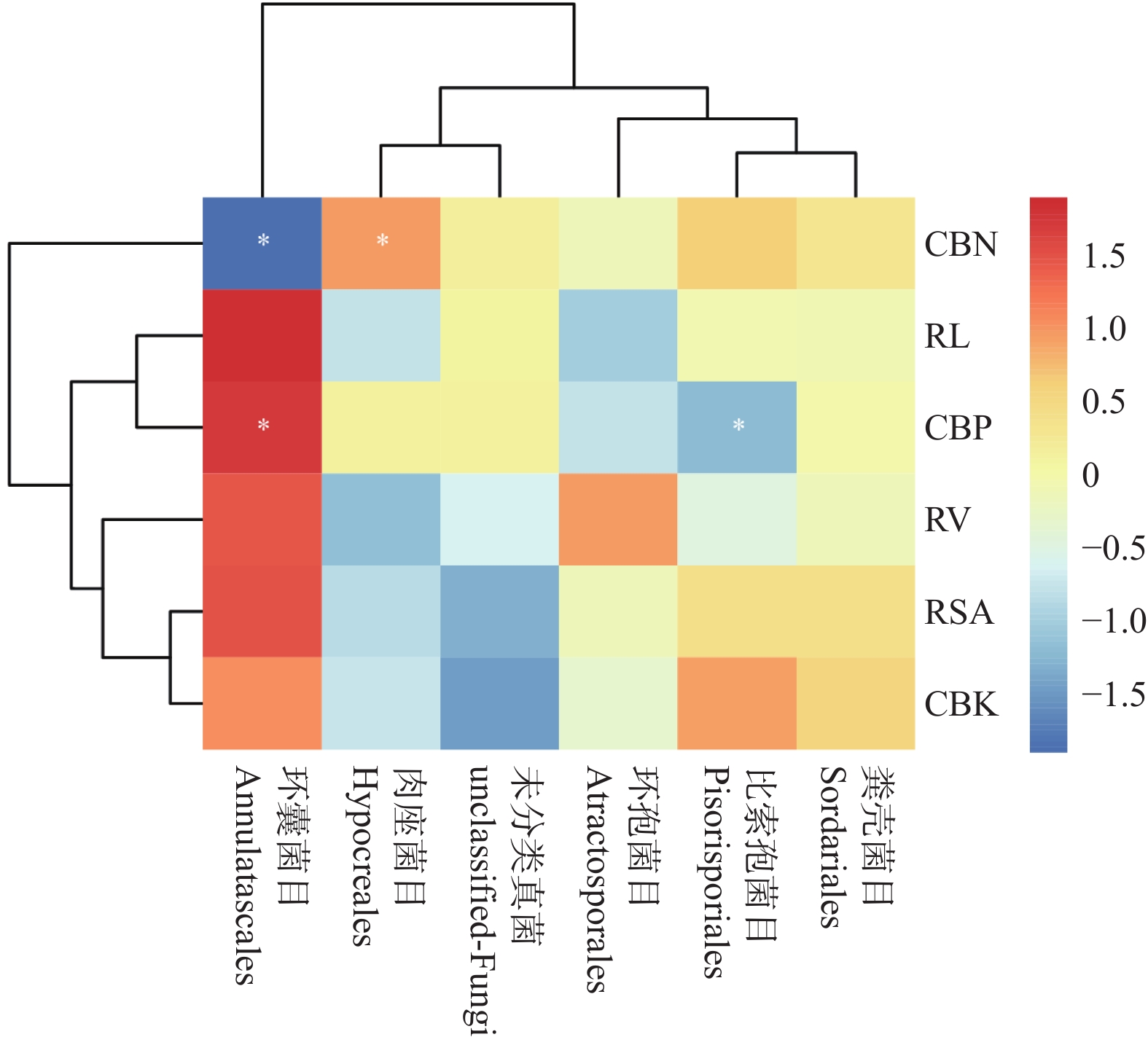
The effects of different nitrogen treatments on root growth and fungal community structures in both the roots and rhizosphere of budded seedling of rubber tree (Hevea brasiliensis) were investigated to determine optimal nitrogen application rate for robust growth of the budded seedlings. Mini-seedling buddings of Clone Reyan 7-33-97 of rubber tree at the age of 3 months old were selected as budded seedlings for experiment and applied with nitrogen fertilizer at different rates. An outdoor pot experiment was conducted with four treatments, control (CK, no nitrogen), low nitrogen (LN, 0.32 g·kg−1), medium nitrogen (MN, 0.64 g·kg−1), and high nitrogen (HN, 1.28 g·kg−1), and root morphological parameters and compositional and diversity shifts in fungal communities in the roots and rhizosphere were determined and analyzed. Results revealed significant effect of different nitrogen treatments on species composition in the roots and rhyzosphere of the mini-seedling buddings. For root fungi, the LN treatment maintained dominance of Basidiomycota and Thelephorales, whereas the HN treatment substantially increased Ascomycota and Sordariales; in the rhizosphere, the LN and MN treatments elevated unclassified fungi and Hypocreales, while HN significantly suppressed Basidiomycota and Annulatascales. The LN treatment (0.32 g·kg−1) maximized alpha diversity, with root fungal Shannon (3.22) and Simpson (0.904) indices increasing by 15.84% and 12.26% respectively as against the CK, while rhizosphere Shannon (3.13) and Simpson (0.914) indices rose by 18.11% and 16.46%, respectively. Root Chao1 (385.97) and ACE (391.43) indices peaked in the LN treatment, 1.16-fold and 1.27-fold higher than that of CK; rhizosphere Chao1 and ACE indices in the LN treatment still elevated significantly, 72.01% and 68.61% higher than those of CK. Beta diversity exhibited highly significant differences in the roots and rhyzosphere among nitrogen treatments and application location, with higher difference in the rhizosphere than in the roots among the nitrogen treatments. Principal Co-ordinates Analysis demonstrated convergent clustering among nitrogen treatments (LN-HN), which was distinctly separated from CK, and higher nitrogen sensitivity in rhizosphere fungi than in the root fungi (R2 difference: 6.07%). The LN treatment (0.32 g·kg−1) was hence recommended as it helps enhance the diversity and richness of the fungal community in both the root system and the rhizosphere, thereby optimizing the fungal community structure in the rhizosphere of mini-seedling buddings.
The effects of different nitrogen treatments on root growth and fungal community structures in both the roots and rhizosphere of budded seedling of rubber tree (Hevea brasiliensis) were investigated to determine optimal nitrogen application rate for robust growth of the budded seedlings. Mini-seedling buddings of Clone Reyan 7-33-97 of rubber tree at the age of 3 months old were selected as budded seedlings for experiment and applied with nitrogen fertilizer at different rates. An outdoor pot experiment was conducted with four treatments, control (CK, no nitrogen), low nitrogen (LN, 0.32 g·kg−1), medium nitrogen (MN, 0.64 g·kg−1), and high nitrogen (HN, 1.28 g·kg−1), and root morphological parameters and compositional and diversity shifts in fungal communities in the roots and rhizosphere were determined and analyzed. Results revealed significant effect of different nitrogen treatments on species composition in the roots and rhyzosphere of the mini-seedling buddings. For root fungi, the LN treatment maintained dominance of Basidiomycota and Thelephorales, whereas the HN treatment substantially increased Ascomycota and Sordariales; in the rhizosphere, the LN and MN treatments elevated unclassified fungi and Hypocreales, while HN significantly suppressed Basidiomycota and Annulatascales. The LN treatment (0.32 g·kg−1) maximized alpha diversity, with root fungal Shannon (3.22) and Simpson (0.904) indices increasing by 15.84% and 12.26% respectively as against the CK, while rhizosphere Shannon (3.13) and Simpson (0.914) indices rose by 18.11% and 16.46%, respectively. Root Chao1 (385.97) and ACE (391.43) indices peaked in the LN treatment, 1.16-fold and 1.27-fold higher than that of CK; rhizosphere Chao1 and ACE indices in the LN treatment still elevated significantly, 72.01% and 68.61% higher than those of CK. Beta diversity exhibited highly significant differences in the roots and rhyzosphere among nitrogen treatments and application location, with higher difference in the rhizosphere than in the roots among the nitrogen treatments. Principal Co-ordinates Analysis demonstrated convergent clustering among nitrogen treatments (LN-HN), which was distinctly separated from CK, and higher nitrogen sensitivity in rhizosphere fungi than in the root fungi (R2 difference: 6.07%). The LN treatment (0.32 g·kg−1) was hence recommended as it helps enhance the diversity and richness of the fungal community in both the root system and the rhizosphere, thereby optimizing the fungal community structure in the rhizosphere of mini-seedling buddings.
2026, 17(3): 451-462.
doi: 10.15886/j.cnki.rdswxb.20240204
Abstract:
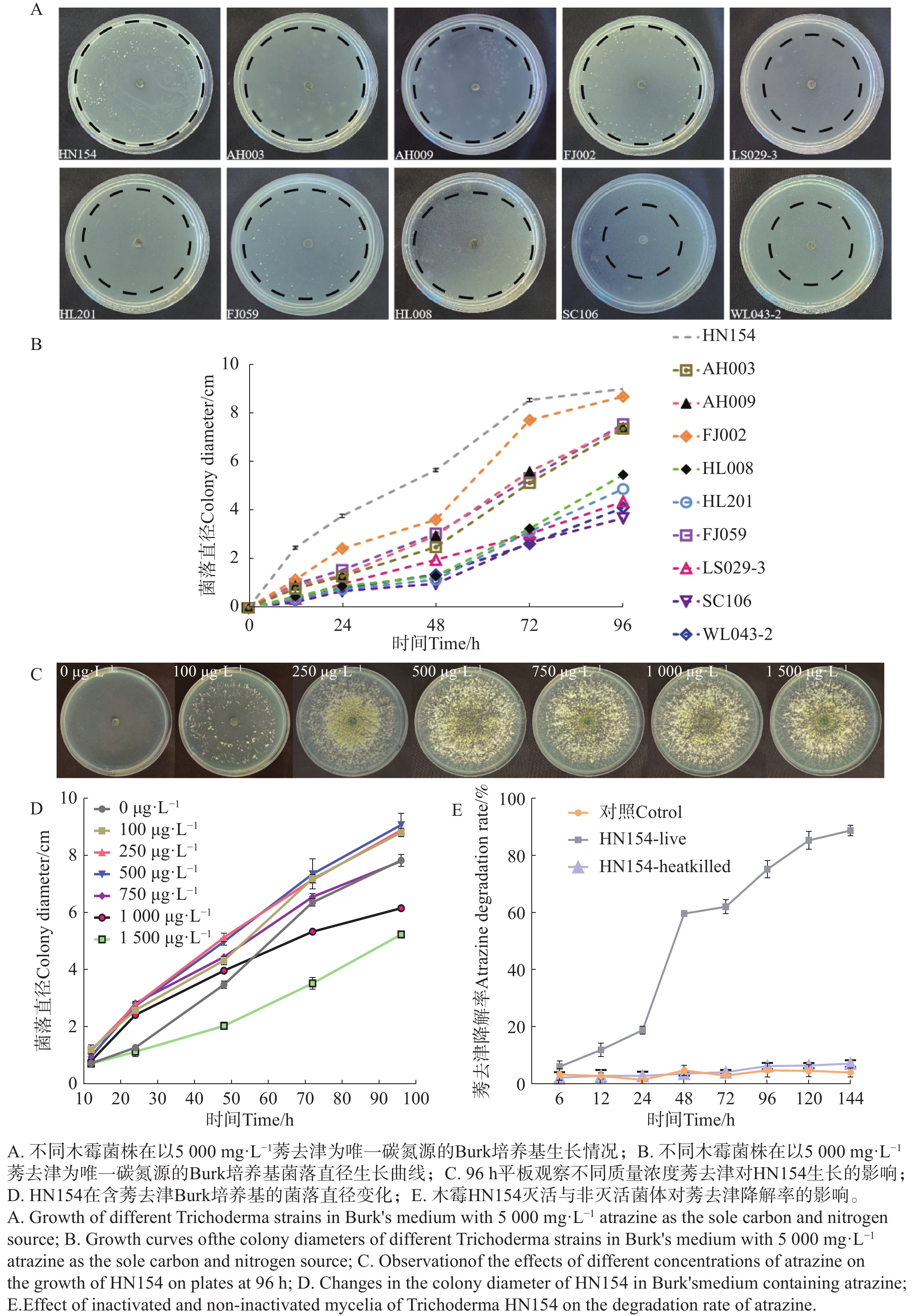
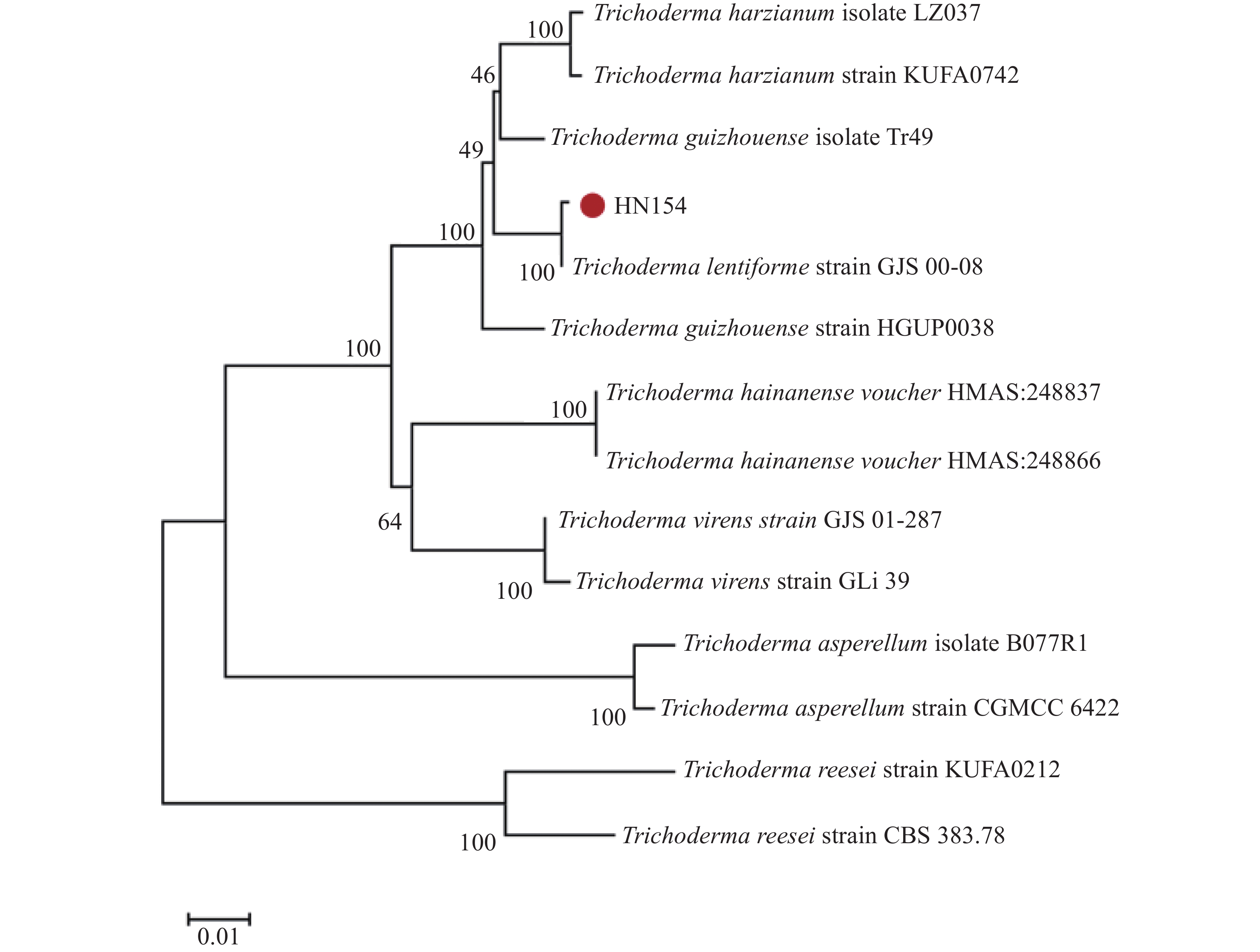
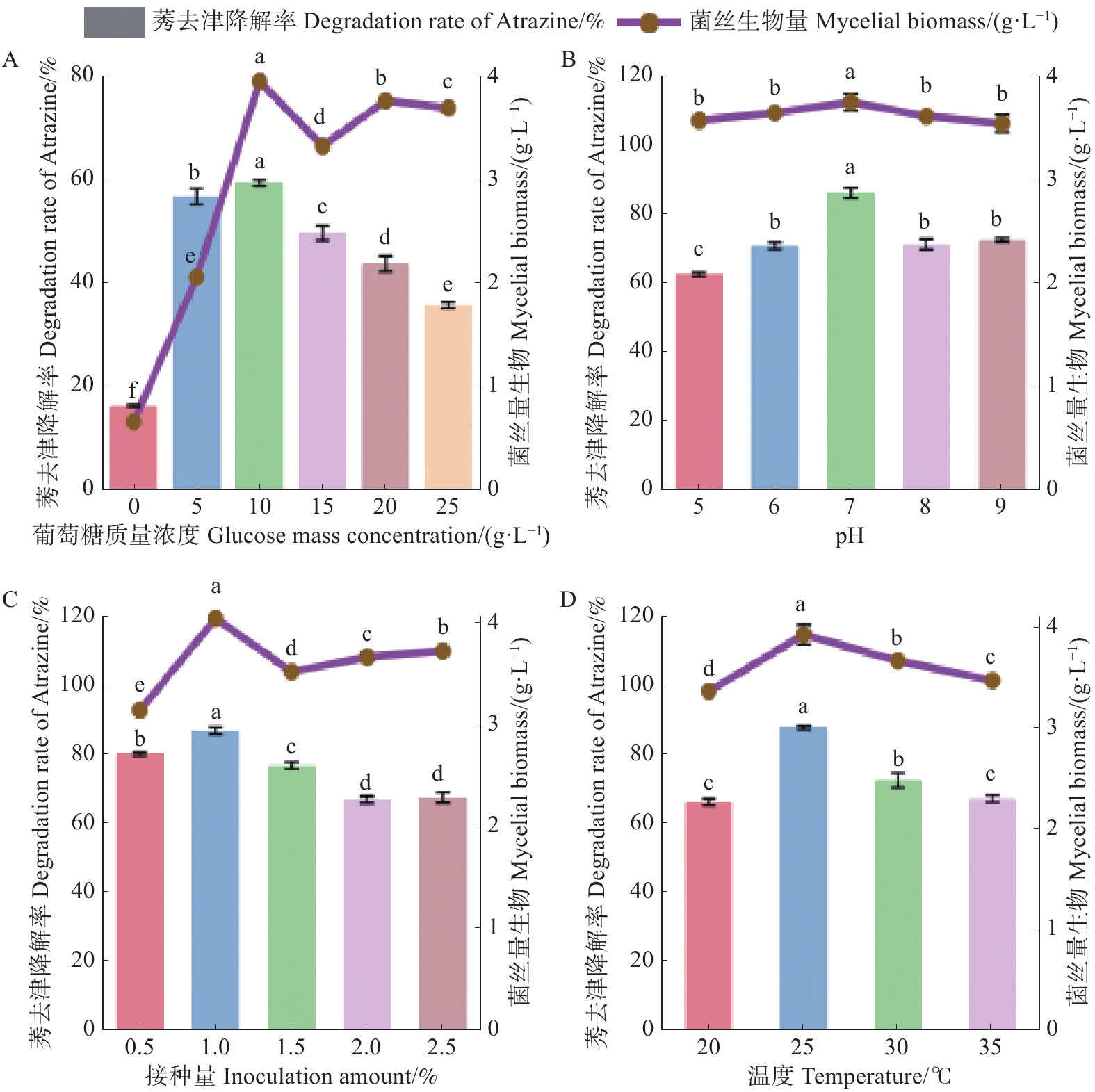
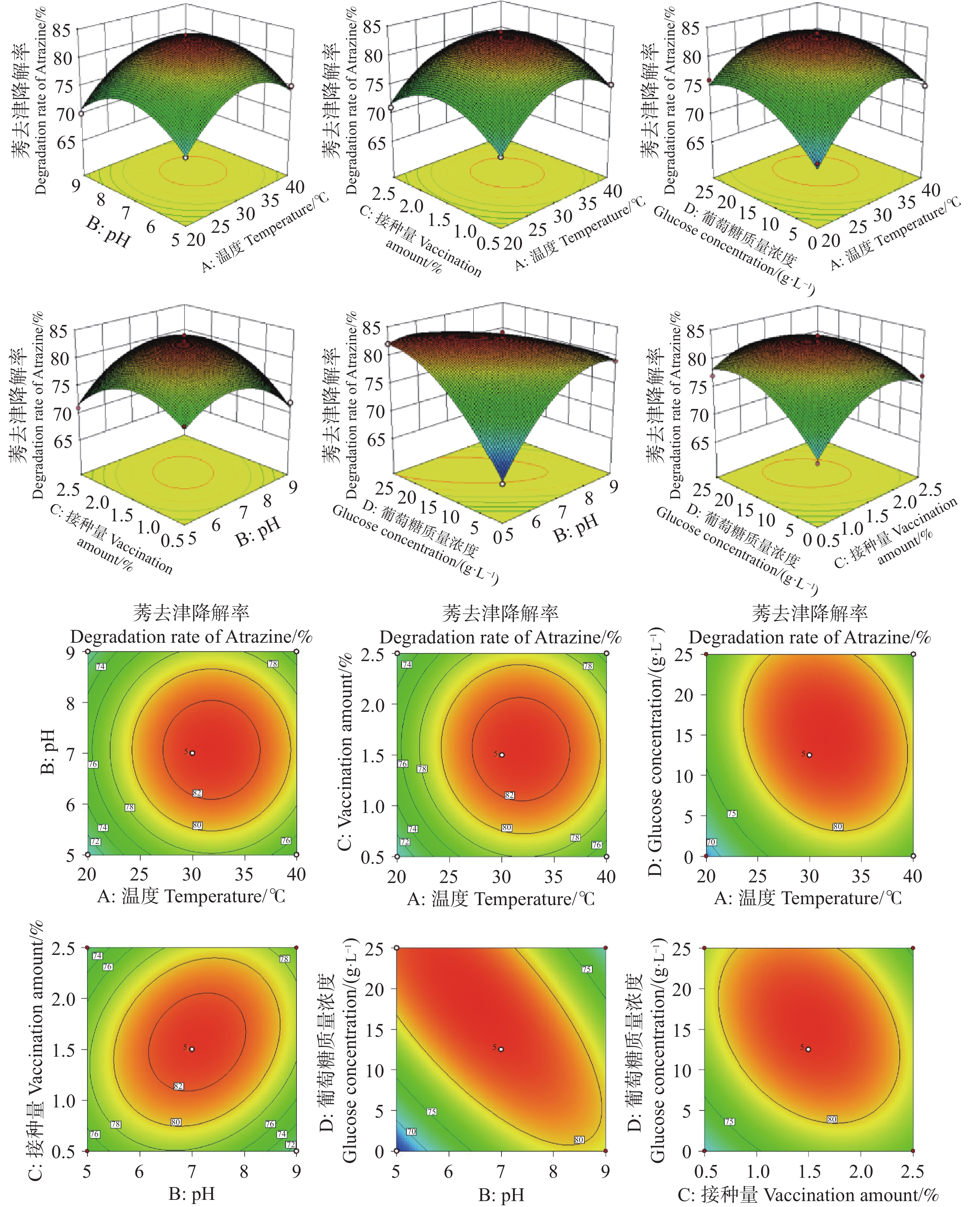
The inorganic salt medium with atrazine as the sole carbon and nitrogen source was used to screen a Trichoderma strain with the best growth performance among 10 Trichoderma strains, and the Trichoderma strain HN154 was selected. The degradation test of atrazine demonstrated that this strain had a high ability to degrade atrazine. Strain HN154 was identified as Trichoderma lentiforme based on morphological characteristics and ITS/RPB2 gene sequence analysis. Optimization of atrazine degradation conditions showed that the atrazine degradation rate was as high as 87.79% when cultured in inorganic salt medium with 10 g·L−1 glucose, pH7, 25 ℃, and 1% inoculum. Response surface optimization further improved degradation to 89.02% under the conditions of 5.24 g·L−1 glucose, pH7.78, 0.77% inoculum, and 25.5 ℃. This study provides microbial resources and technical support for efficient atrazine degradation, offering insights into degradation of microbial pesticide residues.
The inorganic salt medium with atrazine as the sole carbon and nitrogen source was used to screen a Trichoderma strain with the best growth performance among 10 Trichoderma strains, and the Trichoderma strain HN154 was selected. The degradation test of atrazine demonstrated that this strain had a high ability to degrade atrazine. Strain HN154 was identified as Trichoderma lentiforme based on morphological characteristics and ITS/RPB2 gene sequence analysis. Optimization of atrazine degradation conditions showed that the atrazine degradation rate was as high as 87.79% when cultured in inorganic salt medium with 10 g·L−1 glucose, pH7, 25 ℃, and 1% inoculum. Response surface optimization further improved degradation to 89.02% under the conditions of 5.24 g·L−1 glucose, pH7.78, 0.77% inoculum, and 25.5 ℃. This study provides microbial resources and technical support for efficient atrazine degradation, offering insights into degradation of microbial pesticide residues.

2026, 17(3): 463-473.
doi: 10.15886/j.cnki.rdswxb.20250016
Abstract:
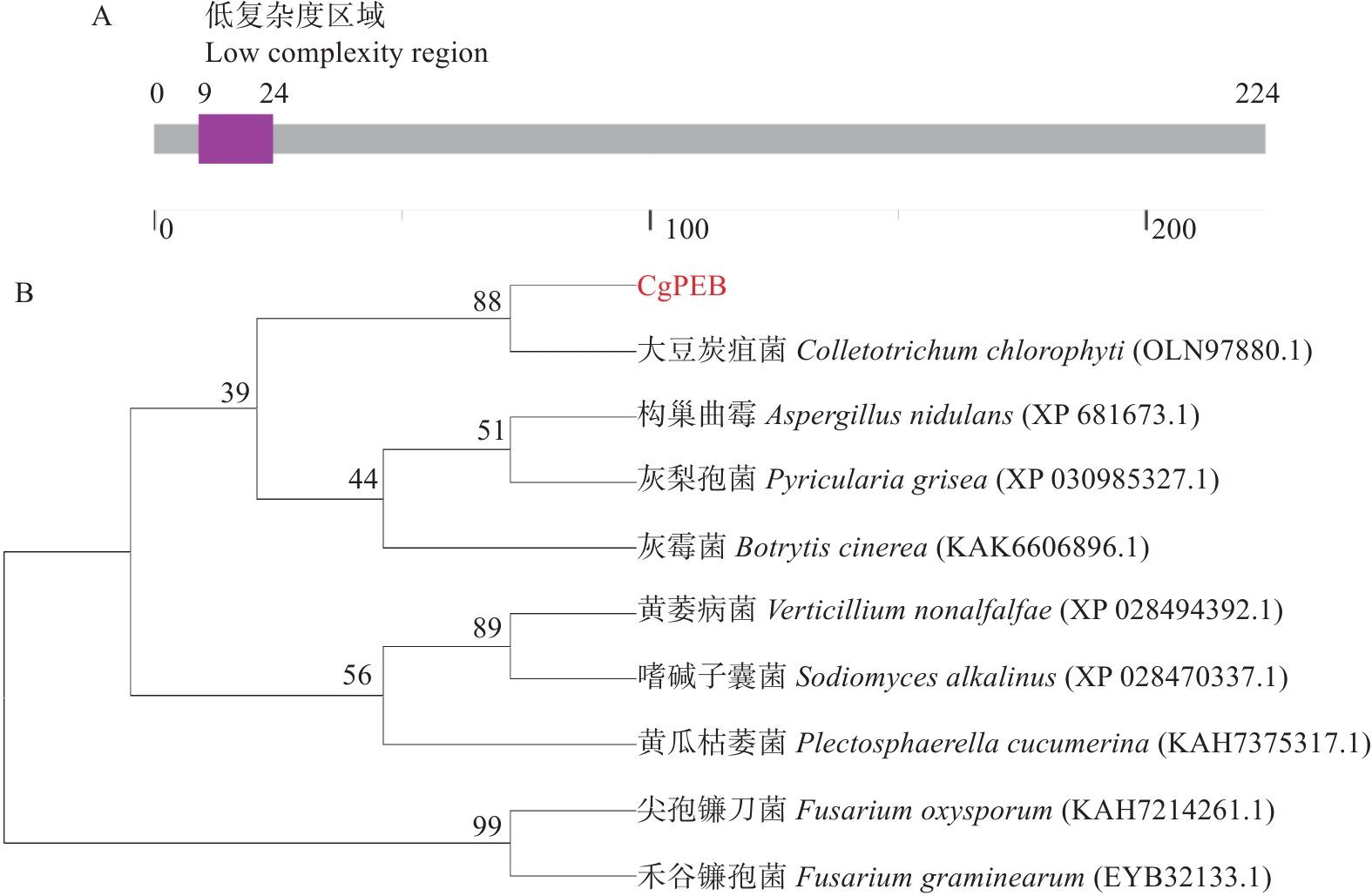
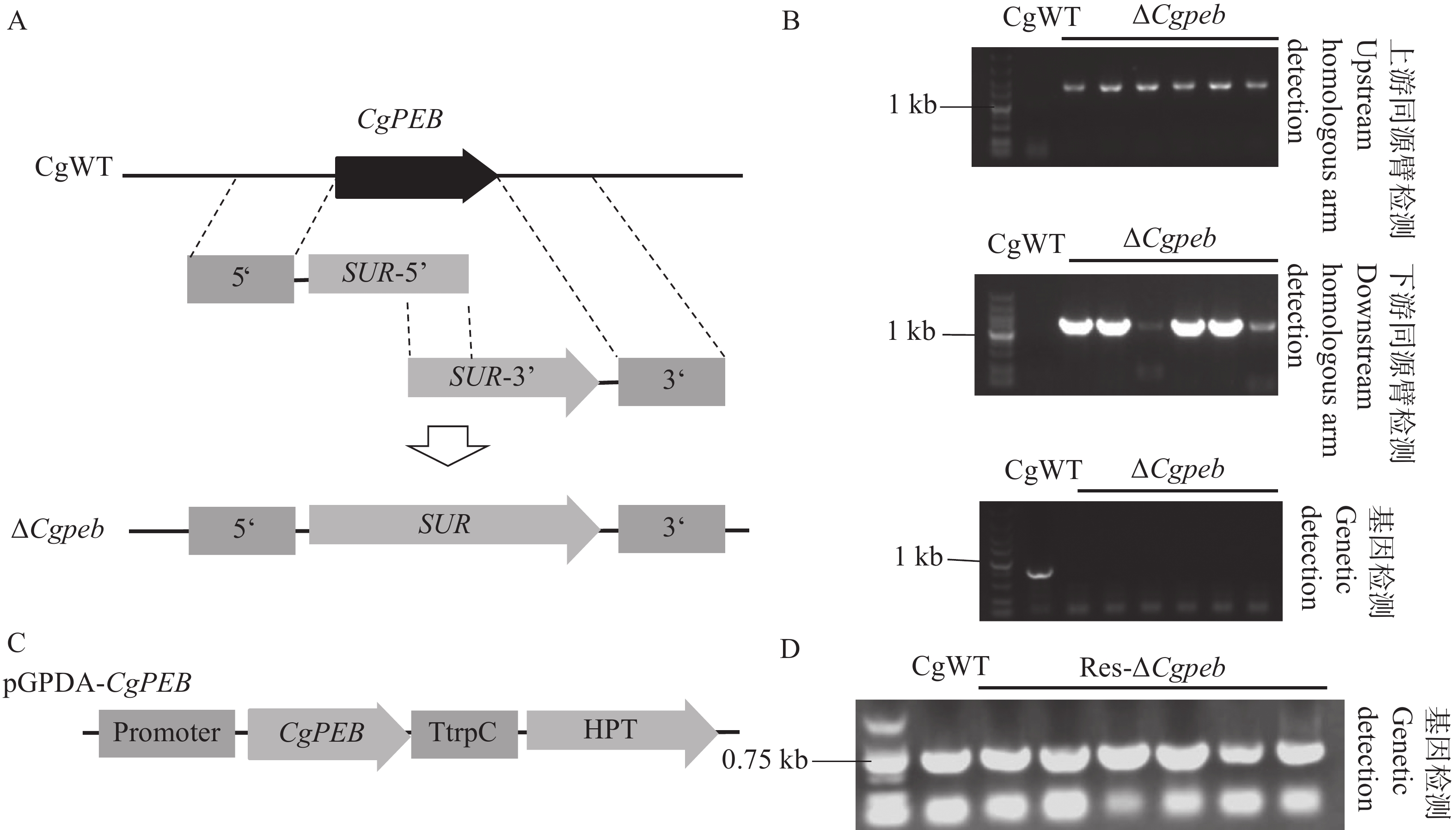
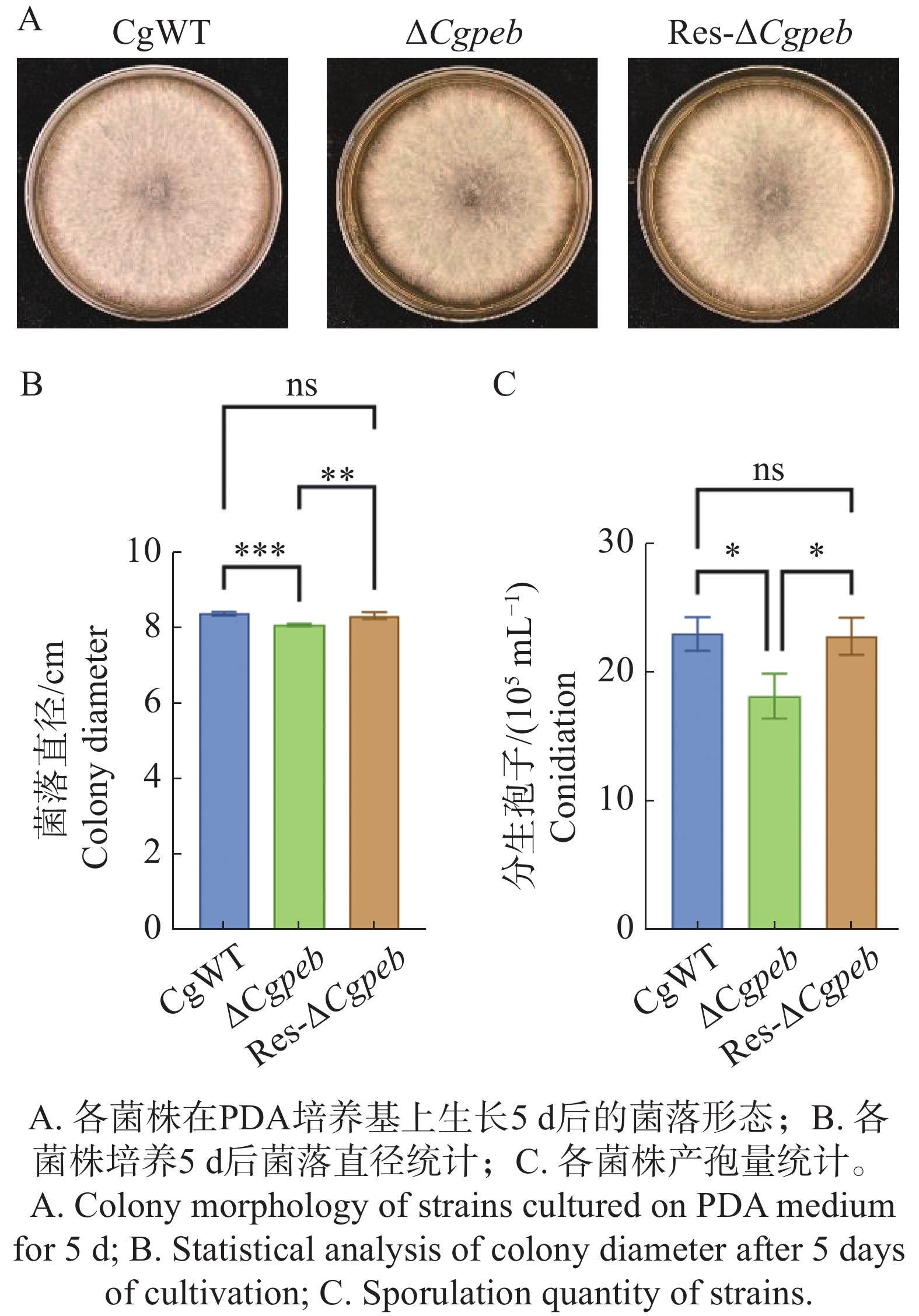
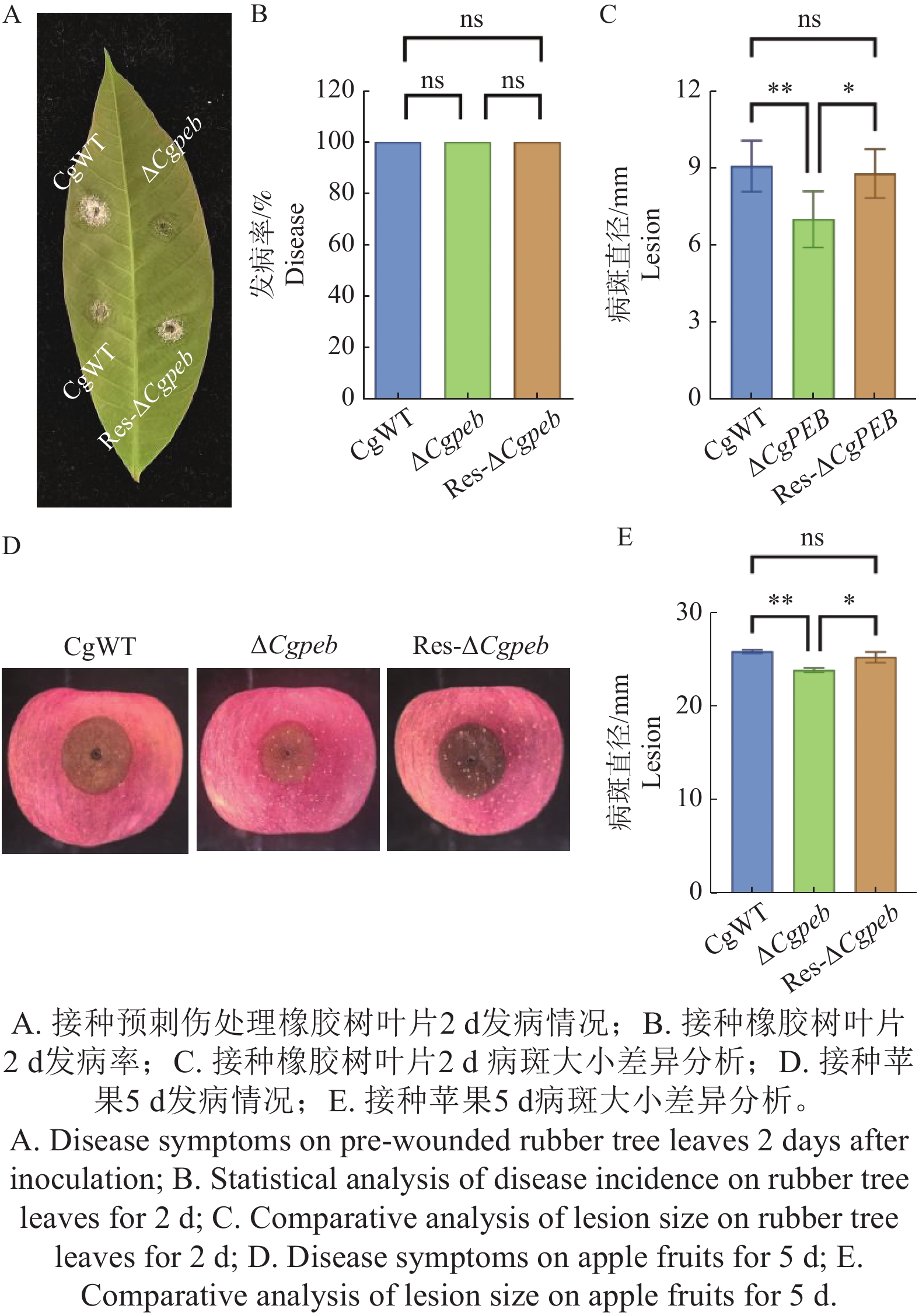
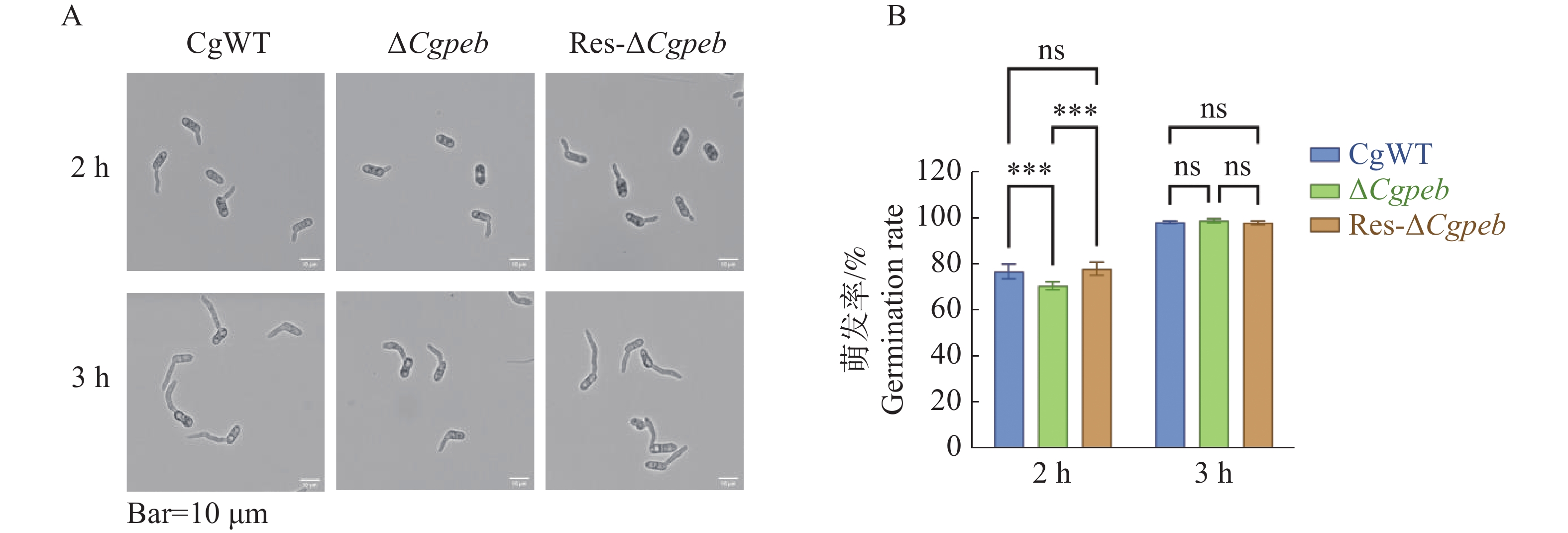
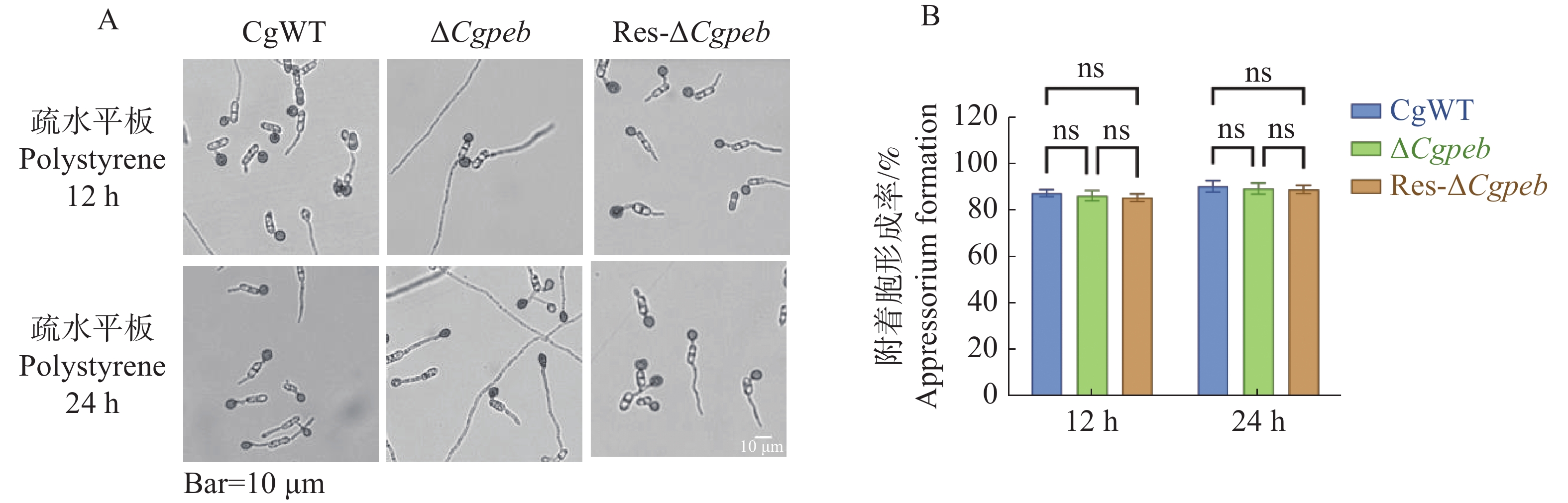
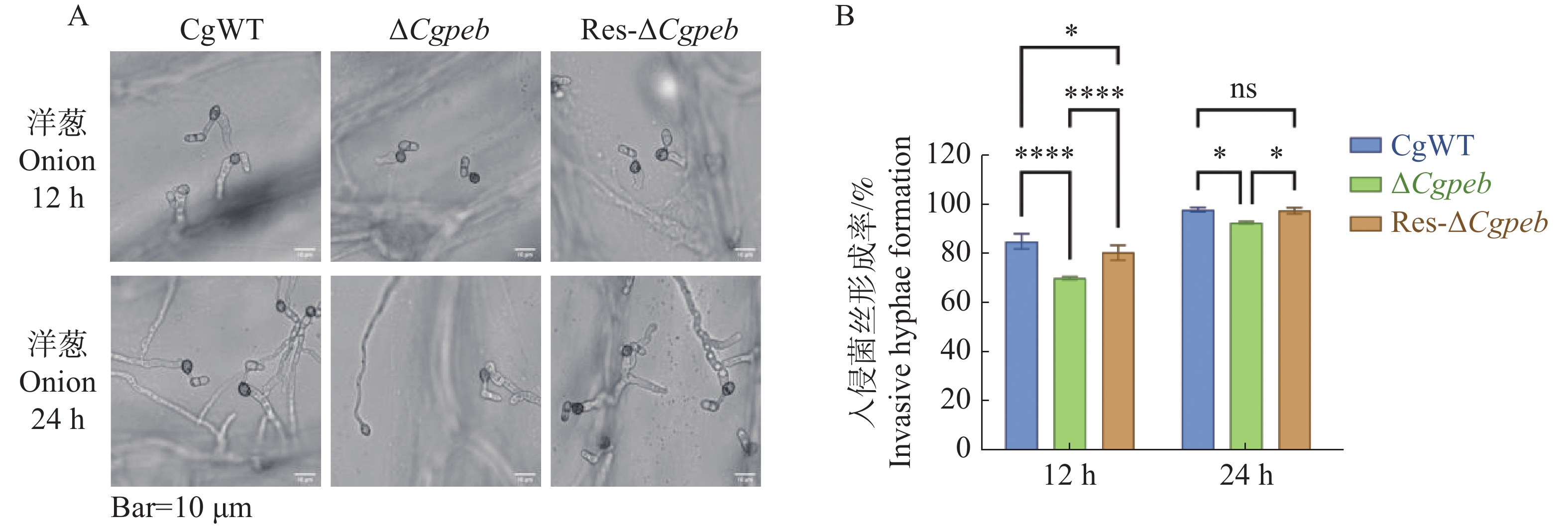
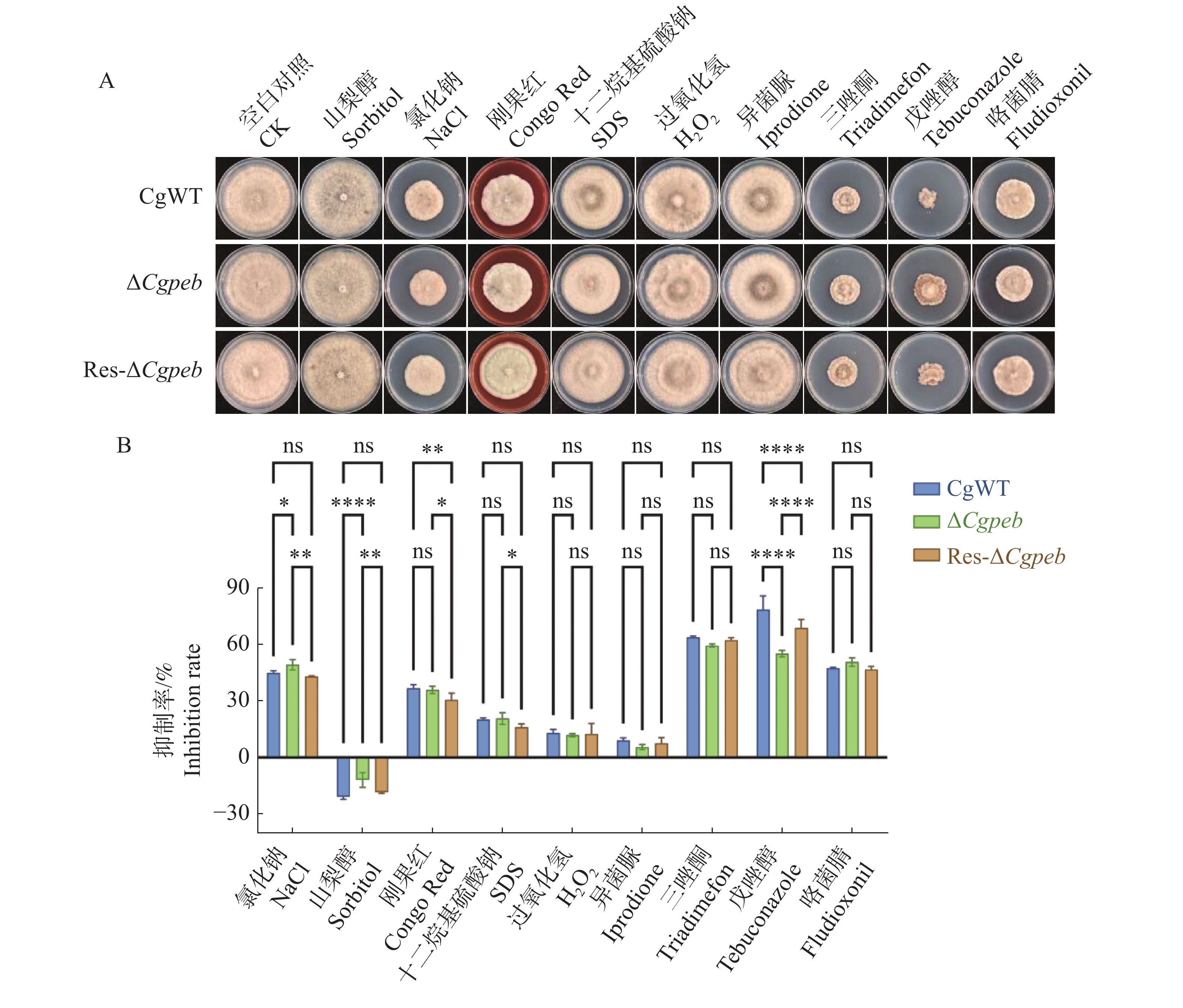
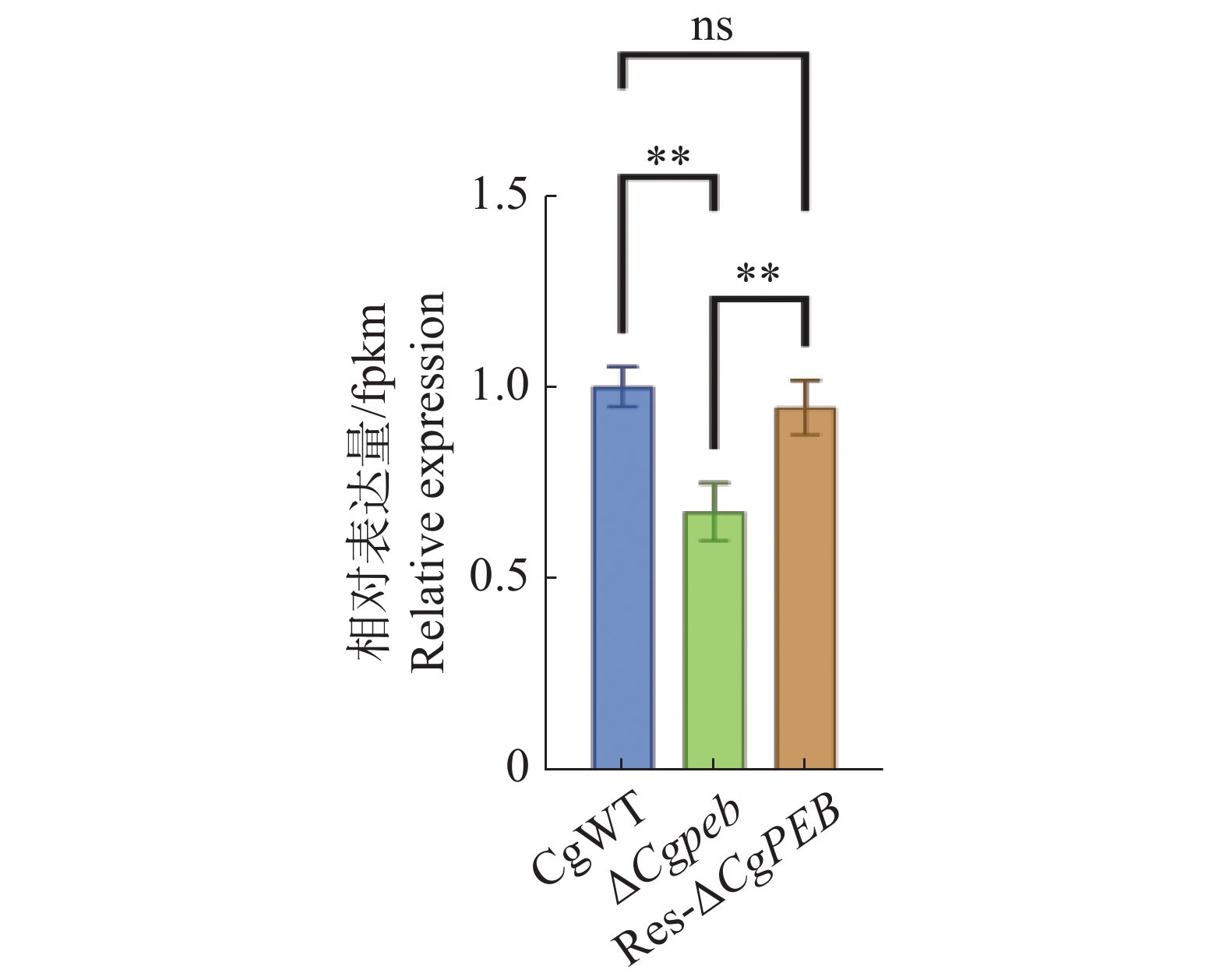
Natural rubber is an important strategic resource in China. The infection of Colletotrichum gloeosporides in rubber tree would greatly affect the development of rubber industry. The functional study of the pathogenicity-related genes of C. gloeosporioides can provide a theoretical basis for disease prevention and control. A gene CgPEB encoding carboxypeptidase Y inhibitor protein was identified in C. gloeosporioides. The gene knockout mutant strain was constructed according to the principle of homologous recombination, and the phenotype and pathogenicity analysis were performed. The results showed that the growth rate and conidial production of ΔCgpeb, Cgpeb gene knockout mutant were reduced. Besides, both the appressorium and invasive hyphae formation rate of ΔCgpeb were significantly reduced. These findings suggested that CgPEB plays an important role in regulating the colony growth, conidial production and pathogenicity of C. gloeosporioides.
Natural rubber is an important strategic resource in China. The infection of Colletotrichum gloeosporides in rubber tree would greatly affect the development of rubber industry. The functional study of the pathogenicity-related genes of C. gloeosporioides can provide a theoretical basis for disease prevention and control. A gene CgPEB encoding carboxypeptidase Y inhibitor protein was identified in C. gloeosporioides. The gene knockout mutant strain was constructed according to the principle of homologous recombination, and the phenotype and pathogenicity analysis were performed. The results showed that the growth rate and conidial production of ΔCgpeb, Cgpeb gene knockout mutant were reduced. Besides, both the appressorium and invasive hyphae formation rate of ΔCgpeb were significantly reduced. These findings suggested that CgPEB plays an important role in regulating the colony growth, conidial production and pathogenicity of C. gloeosporioides.
2026, 17(3): 474-486.
doi: 10.15886/j.cnki.rdswxb.20250041
Abstract:
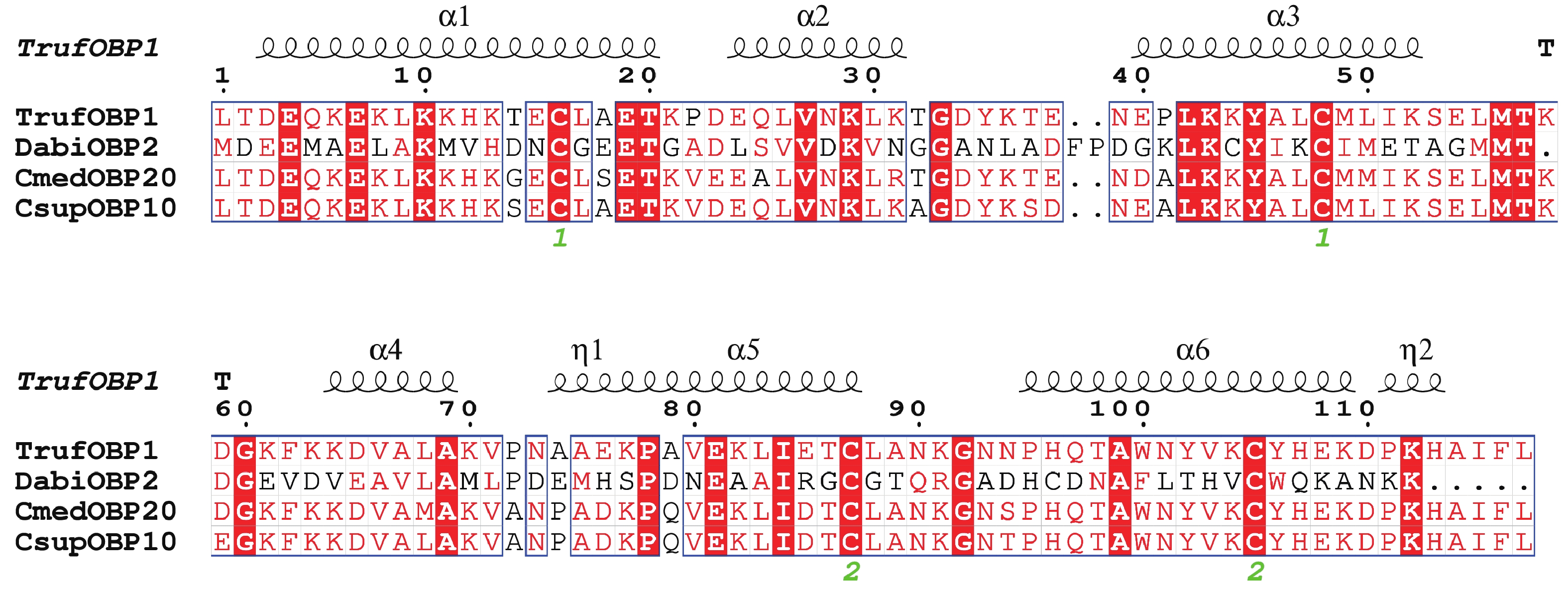
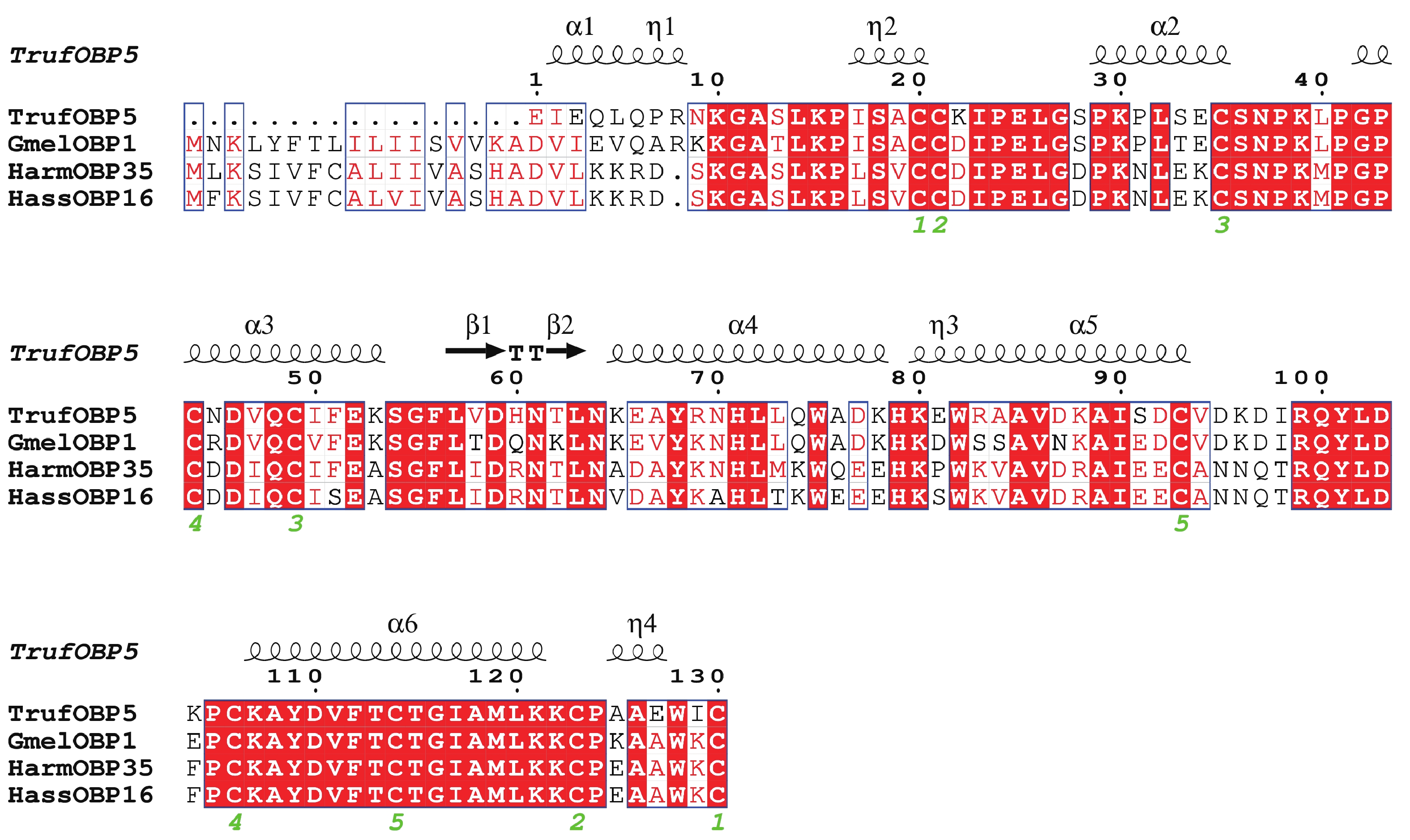
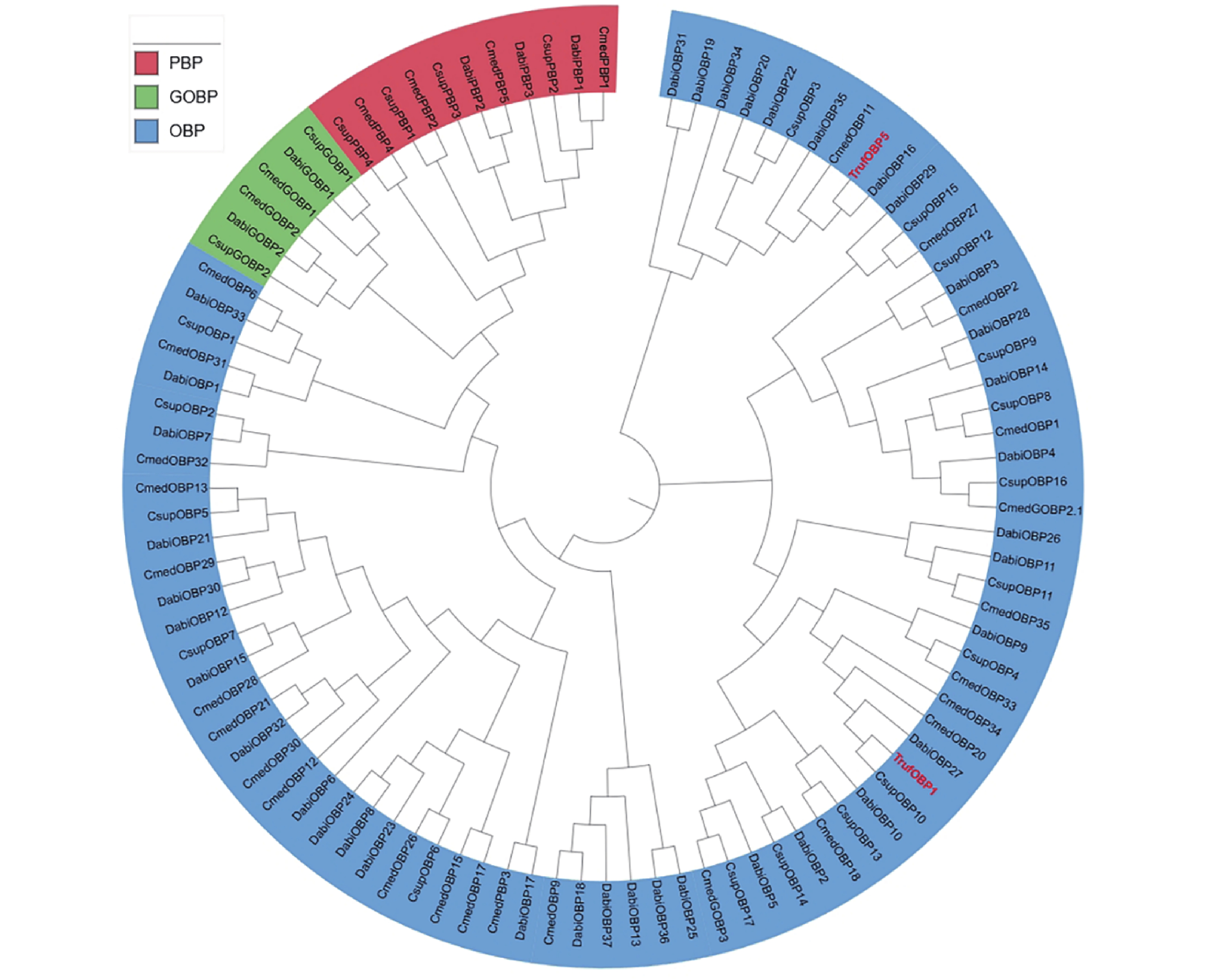
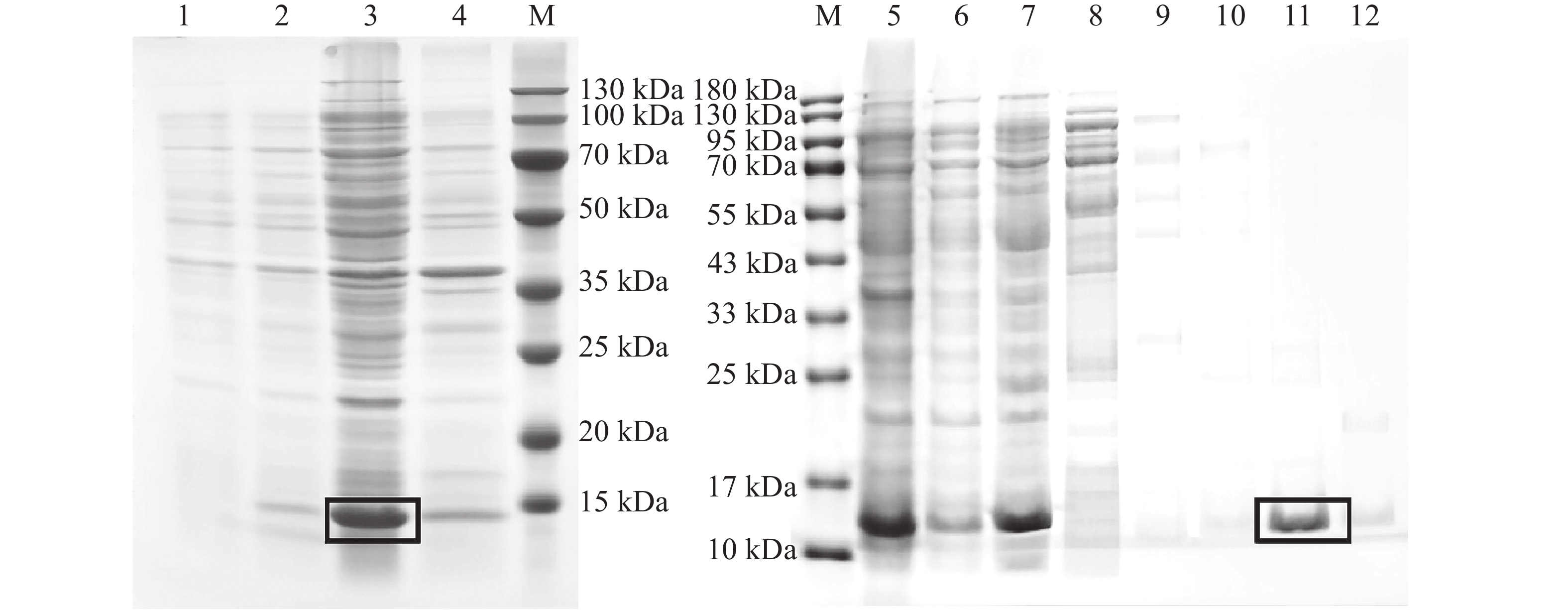
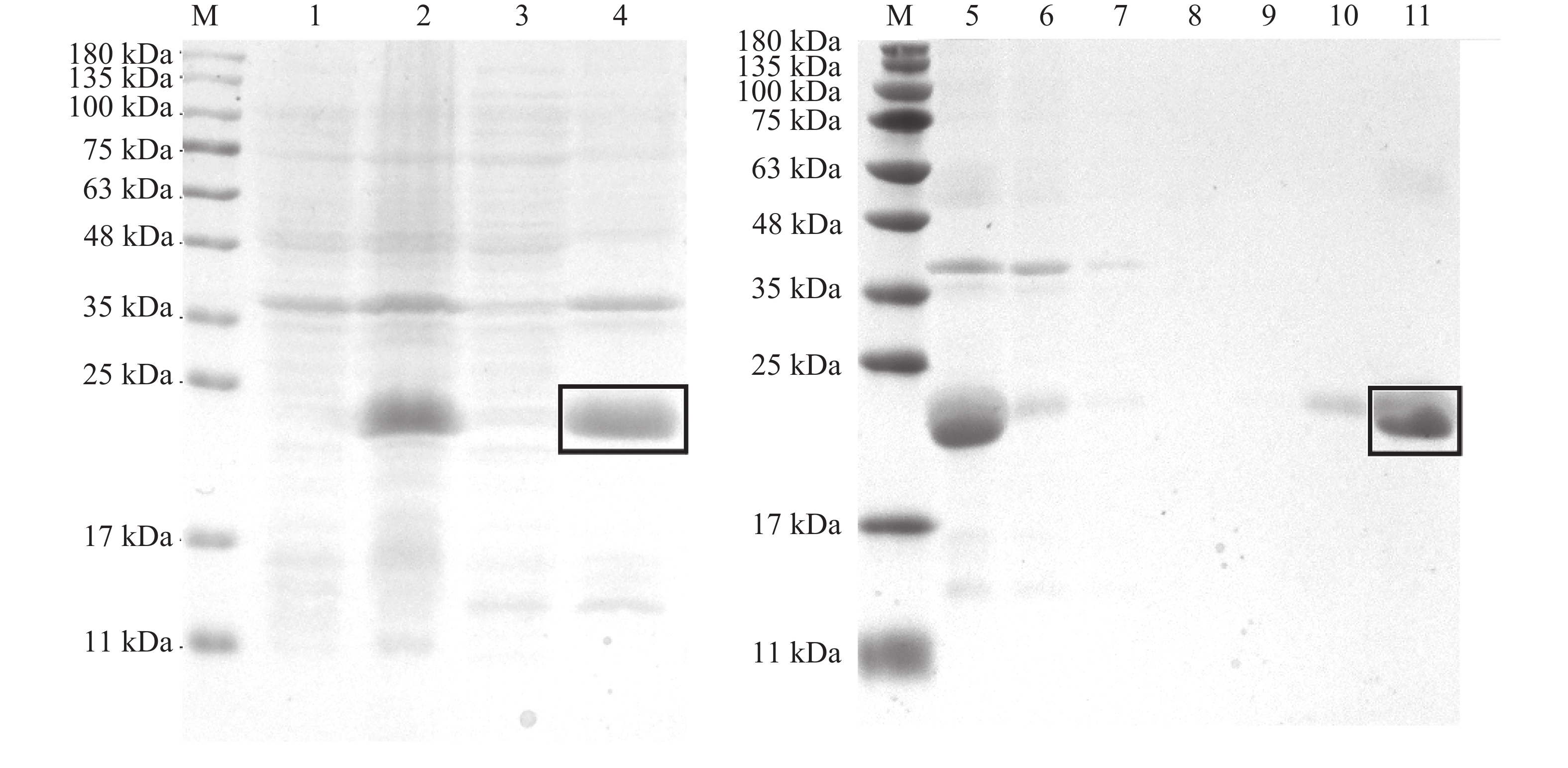
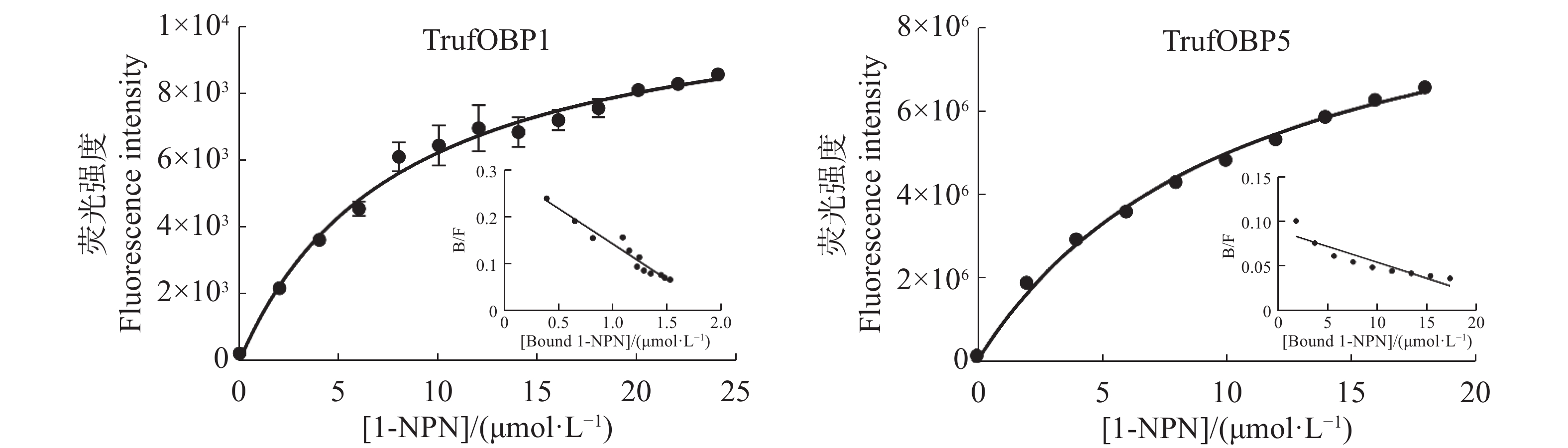
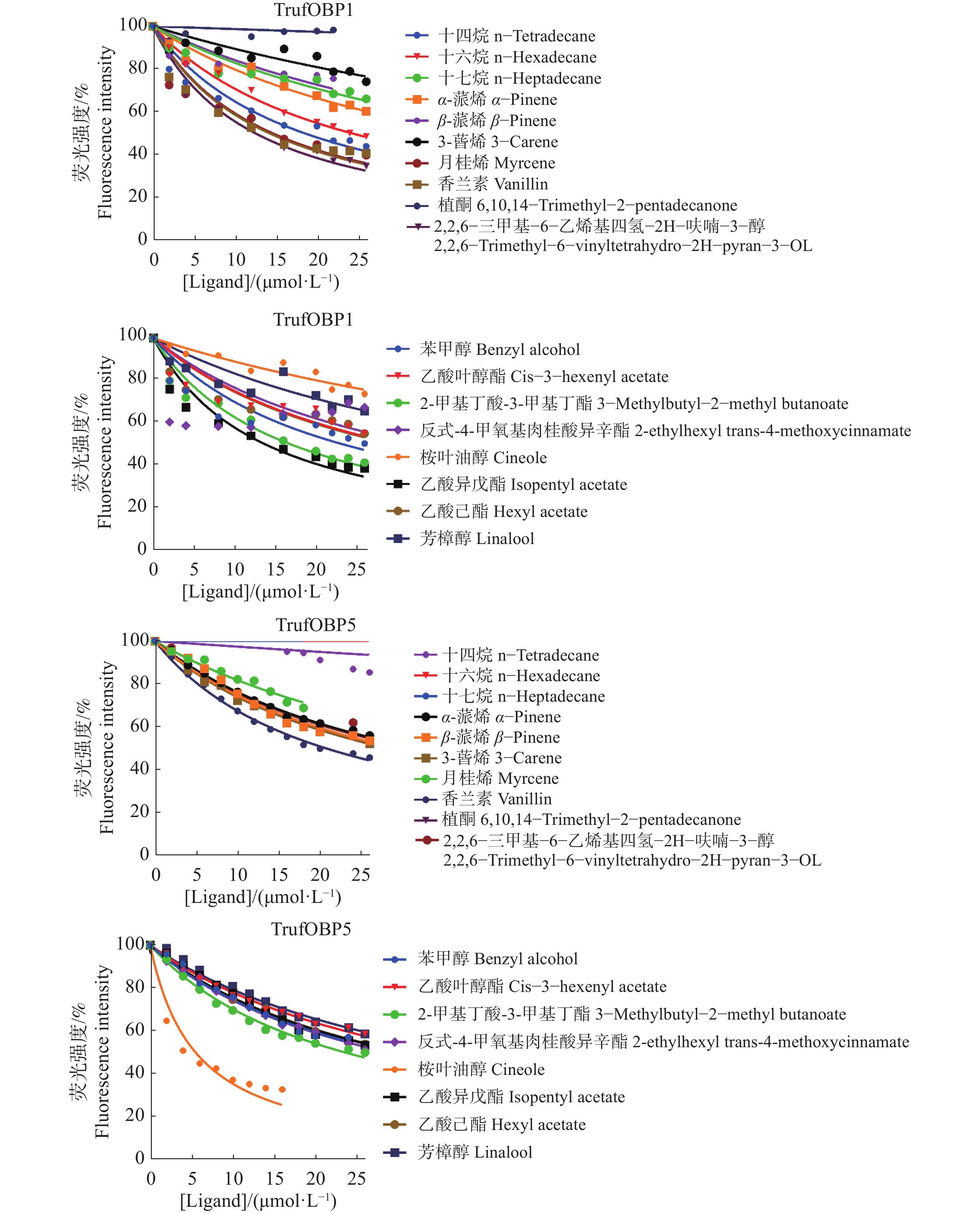
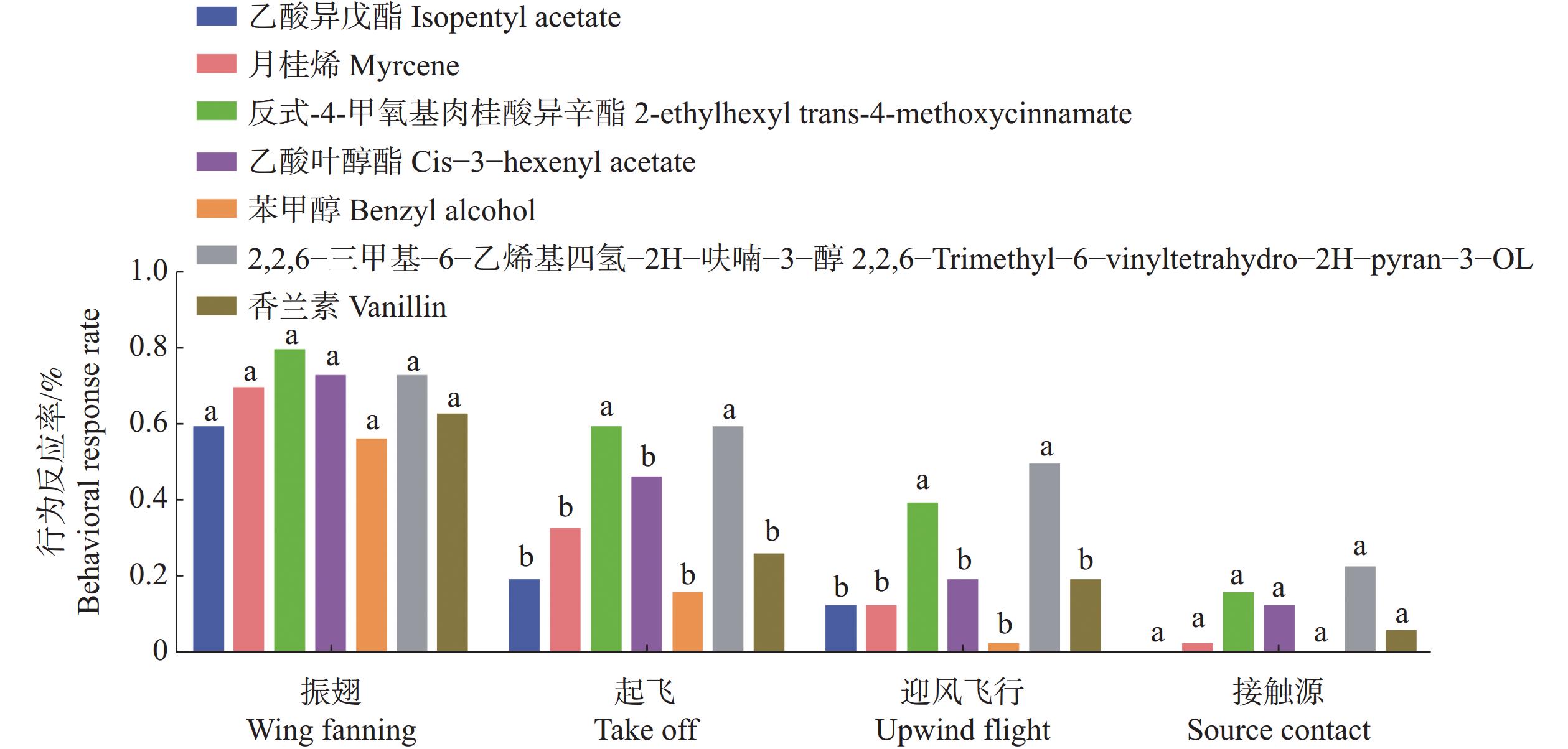
The insect olfactory system plays a pivotal role in host recognition, mating, and oviposition, with odorant-binding proteins (OBPs) serving critical functions in olfactory signal transduction. An attempt was made to analyze the sequence characteristics and ligand-binding properties of two OBPs (TrufOBP1 and TrufOBP5) in Tirathaba rufivena, a major pest infesting inflorescences and fruit of arecanut (Areca catechu), and to validate their behavioral regulatory roles through wind tunnel experiments. Sequence analysis revealed that TrufOBP1 contains four conserved cysteines and is classified as a Minus-C OBP, whereas TrufOBP5 possesses ten conserved cysteines and is categorized as a Plus-C OBP. Fluorescence competitive binding assays demonstrated that TrufOBP1 exhibited broad ligand specificity and was bound to 11 compounds, including nine volatiles from areca nut (Areca catechu) inflorescences and two sex pheromones. In contrast, TrufOBP5 displayed high binding specificity, and interacted exclusively with octyl p-methoxycinnamate and vanillin, suggesting its potential role in host plant recognition. Wind tunnel experiments identified β-myrcene, octyl p-methoxycinnamate, and 3-hexenyl acetate as attractants for female adults from host volatiles. Among sex pheromone components, 2,2,6-trimethyl-6-vinyltetrahydro-2H-pyran-3-OL exhibited the highest attraction activity, while vanillin also demonstrated significant attractant effects. This study elucidates the functional roles of TrufOBP1 and TrufOBP5 in host recognition and sex pheromone perception in T. rufivena, providing a theoretical foundation for pest behavior regulation and supporting the development of olfaction-based green control strategies.
The insect olfactory system plays a pivotal role in host recognition, mating, and oviposition, with odorant-binding proteins (OBPs) serving critical functions in olfactory signal transduction. An attempt was made to analyze the sequence characteristics and ligand-binding properties of two OBPs (TrufOBP1 and TrufOBP5) in Tirathaba rufivena, a major pest infesting inflorescences and fruit of arecanut (Areca catechu), and to validate their behavioral regulatory roles through wind tunnel experiments. Sequence analysis revealed that TrufOBP1 contains four conserved cysteines and is classified as a Minus-C OBP, whereas TrufOBP5 possesses ten conserved cysteines and is categorized as a Plus-C OBP. Fluorescence competitive binding assays demonstrated that TrufOBP1 exhibited broad ligand specificity and was bound to 11 compounds, including nine volatiles from areca nut (Areca catechu) inflorescences and two sex pheromones. In contrast, TrufOBP5 displayed high binding specificity, and interacted exclusively with octyl p-methoxycinnamate and vanillin, suggesting its potential role in host plant recognition. Wind tunnel experiments identified β-myrcene, octyl p-methoxycinnamate, and 3-hexenyl acetate as attractants for female adults from host volatiles. Among sex pheromone components, 2,2,6-trimethyl-6-vinyltetrahydro-2H-pyran-3-OL exhibited the highest attraction activity, while vanillin also demonstrated significant attractant effects. This study elucidates the functional roles of TrufOBP1 and TrufOBP5 in host recognition and sex pheromone perception in T. rufivena, providing a theoretical foundation for pest behavior regulation and supporting the development of olfaction-based green control strategies.

2026, 17(3): 487-497.
doi: 10.15886/j.cnki.rdswxb.20240161
Abstract:
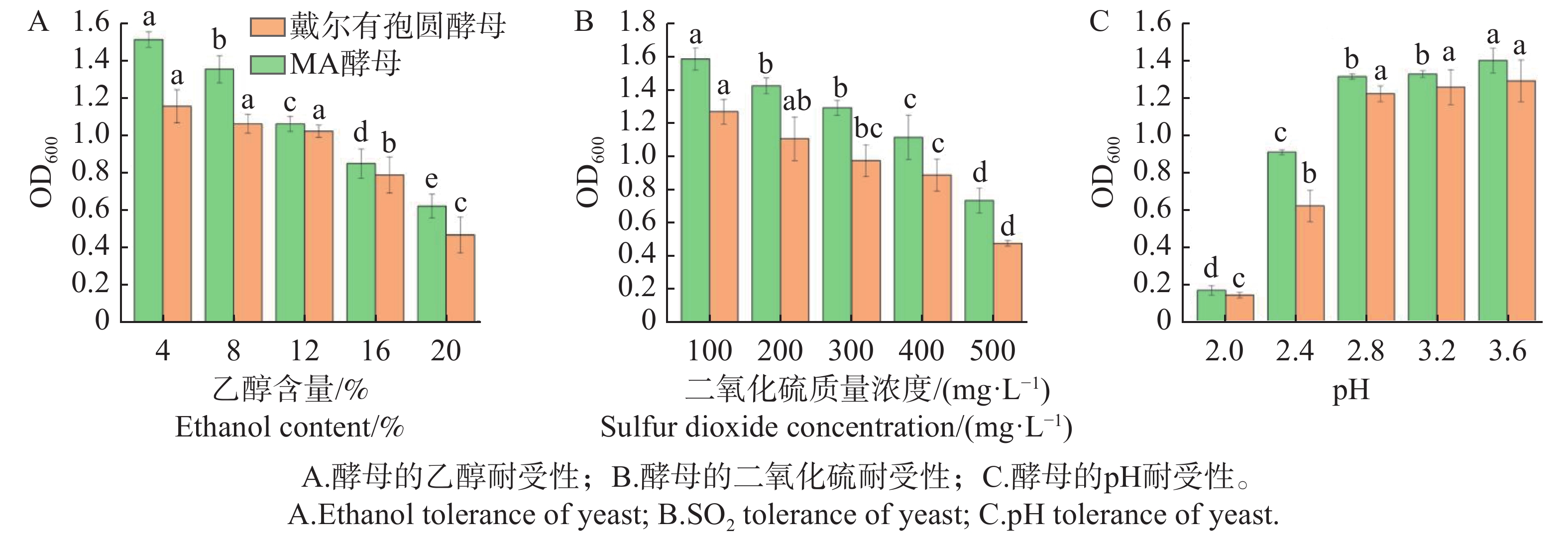
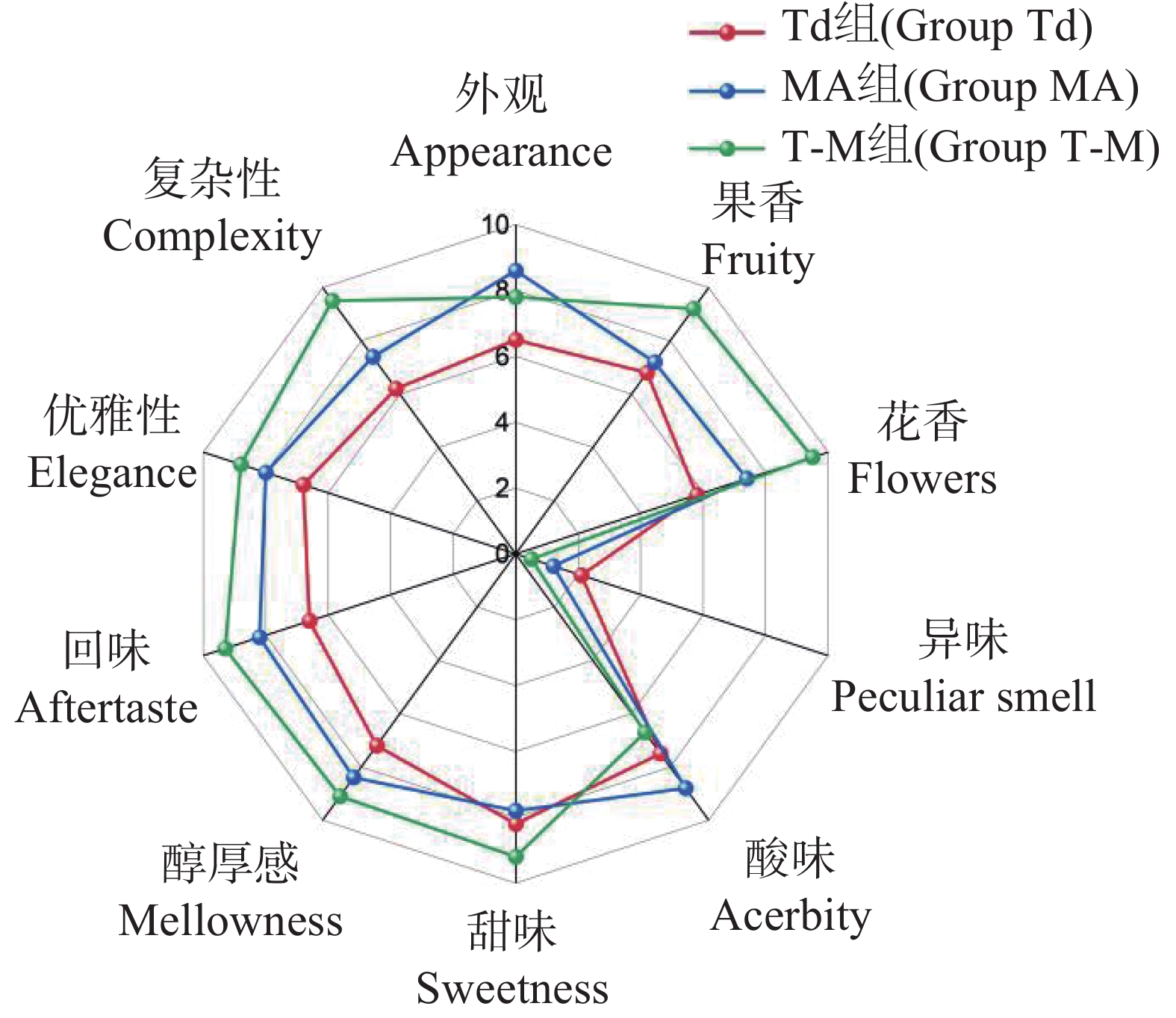
Torulaspora delbrueckii is one of non-Saccharomyces yeasts mostly used for research, and is widely used for brewing of various fruit wines. It is co-fermented with Saccharomyces cerevisiae for improving fruit wine flavors. In this experiment, a wild-type yeast strain was selected from the peel of cantaloupe in Fulo Town, Ledong County, Hainan Province. Through phenotypic observation of the strain and constructing a phylogenetic tree based on 18S rDNA sequencing, the strain was identified as T. delbrueckii. Under the fermentation condition of fruit wine, T. delbrueckii grew normally in the range of alcohol content 4%−20%, pH 2.8−3.6 and sulfur dioxide concentration 100−500 mg·L−1, and the growth rate was the highest under the condition of alcohol content 4%, pH 3.6 and sulfur dioxide concentration 300 mg·L−1. The results show that T. delbrueckii did not produce hydrogen sulfide, had no killing effect on S. cerevisiae, and improved the flavor of cantaloupe fruit wine, which can be used in the production of cantaloupe fruit wine.
Torulaspora delbrueckii is one of non-Saccharomyces yeasts mostly used for research, and is widely used for brewing of various fruit wines. It is co-fermented with Saccharomyces cerevisiae for improving fruit wine flavors. In this experiment, a wild-type yeast strain was selected from the peel of cantaloupe in Fulo Town, Ledong County, Hainan Province. Through phenotypic observation of the strain and constructing a phylogenetic tree based on 18S rDNA sequencing, the strain was identified as T. delbrueckii. Under the fermentation condition of fruit wine, T. delbrueckii grew normally in the range of alcohol content 4%−20%, pH 2.8−3.6 and sulfur dioxide concentration 100−500 mg·L−1, and the growth rate was the highest under the condition of alcohol content 4%, pH 3.6 and sulfur dioxide concentration 300 mg·L−1. The results show that T. delbrueckii did not produce hydrogen sulfide, had no killing effect on S. cerevisiae, and improved the flavor of cantaloupe fruit wine, which can be used in the production of cantaloupe fruit wine.
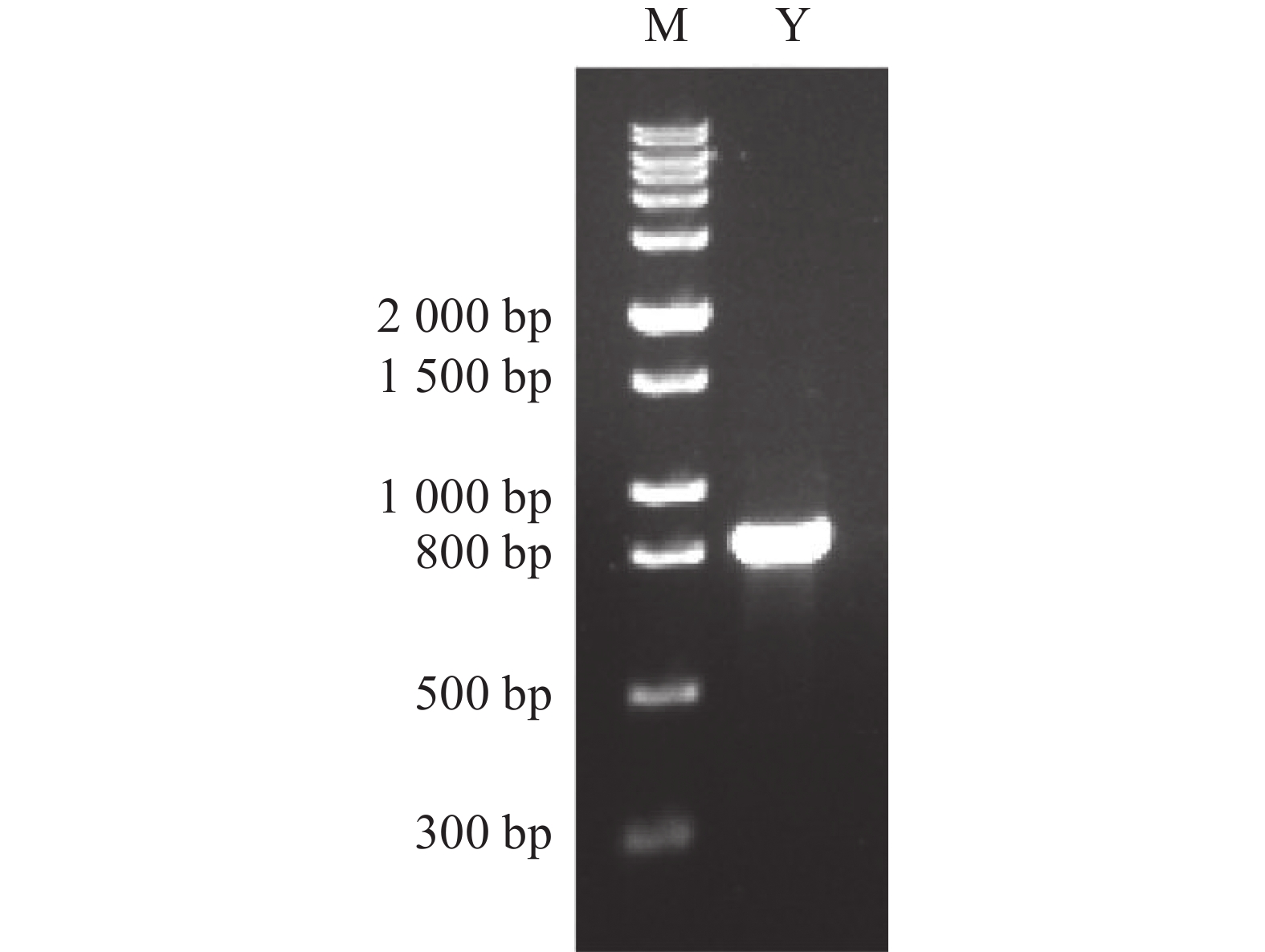
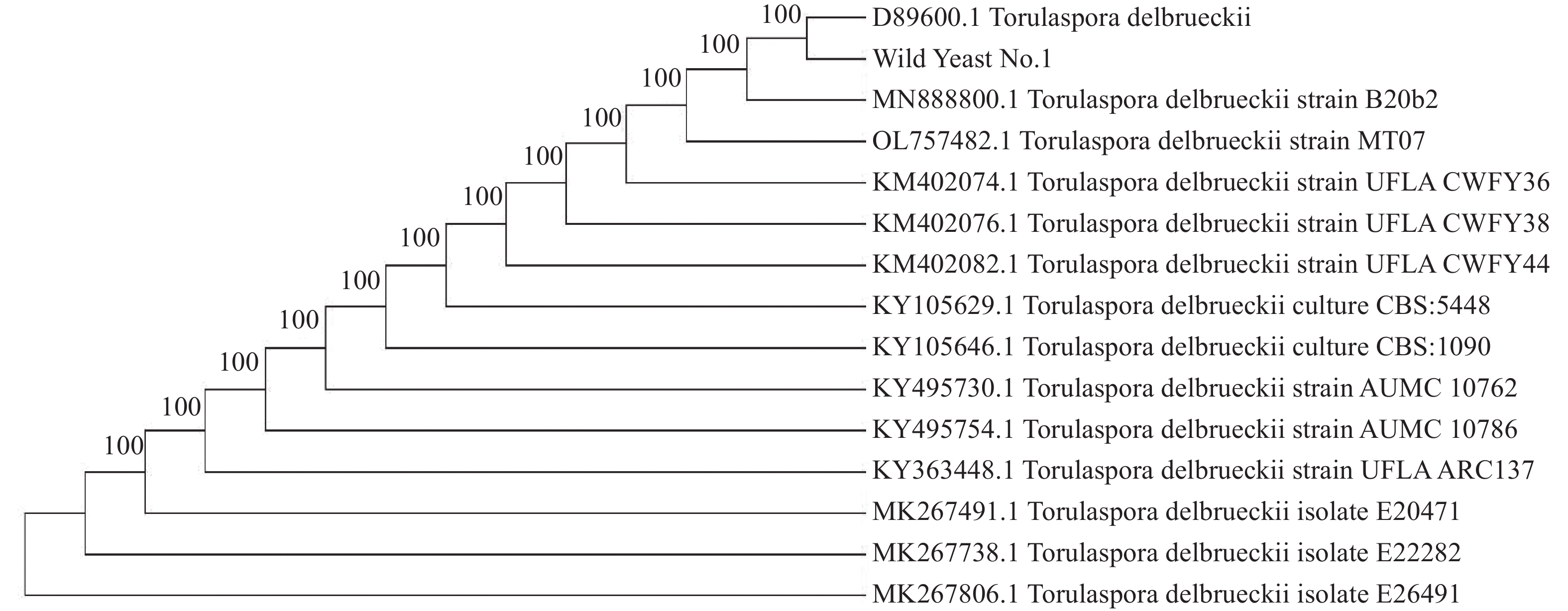
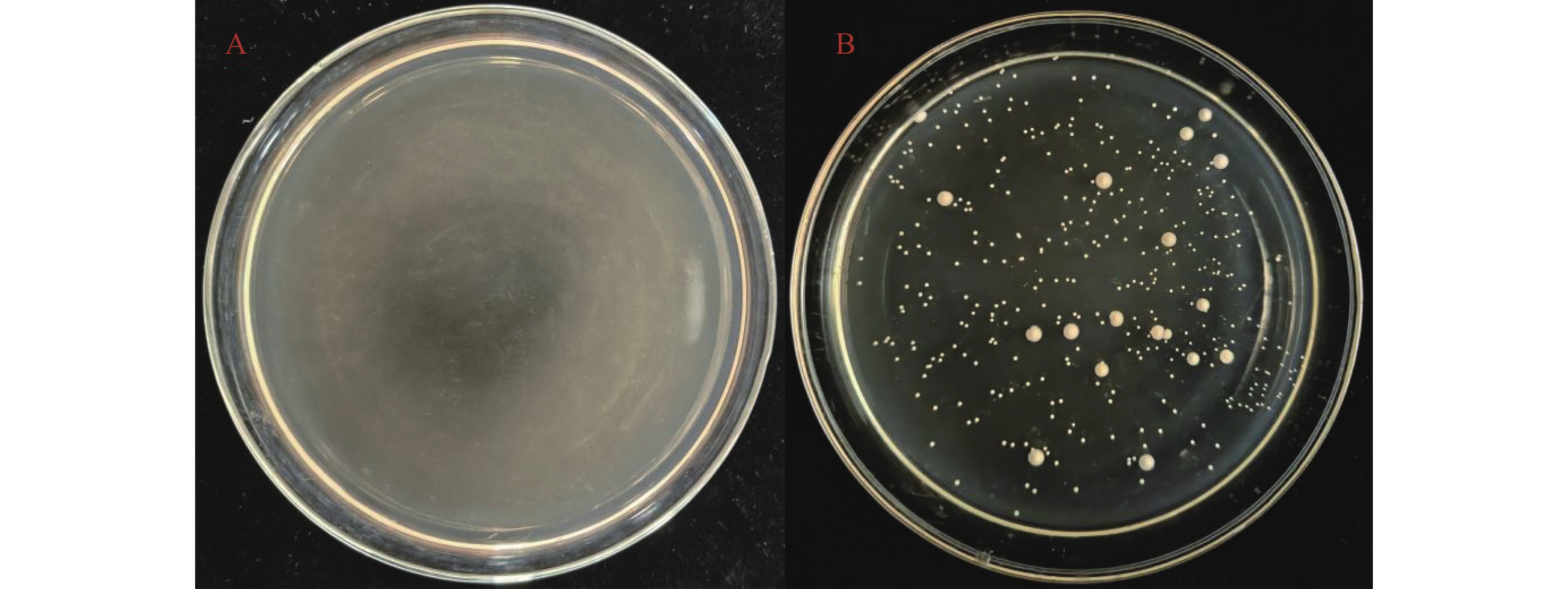
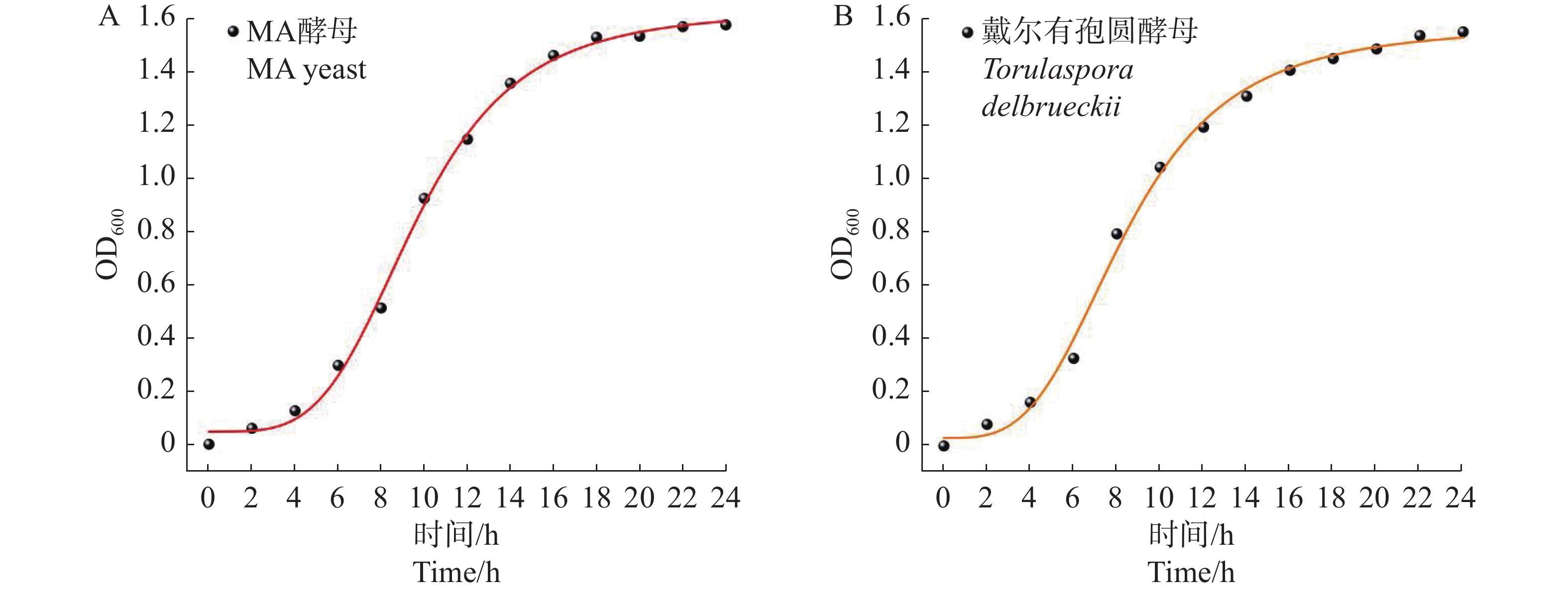
2026, 17(3): 498-507.
doi: 10.15886/j.cnki.rdswxb.20250004
Abstract:
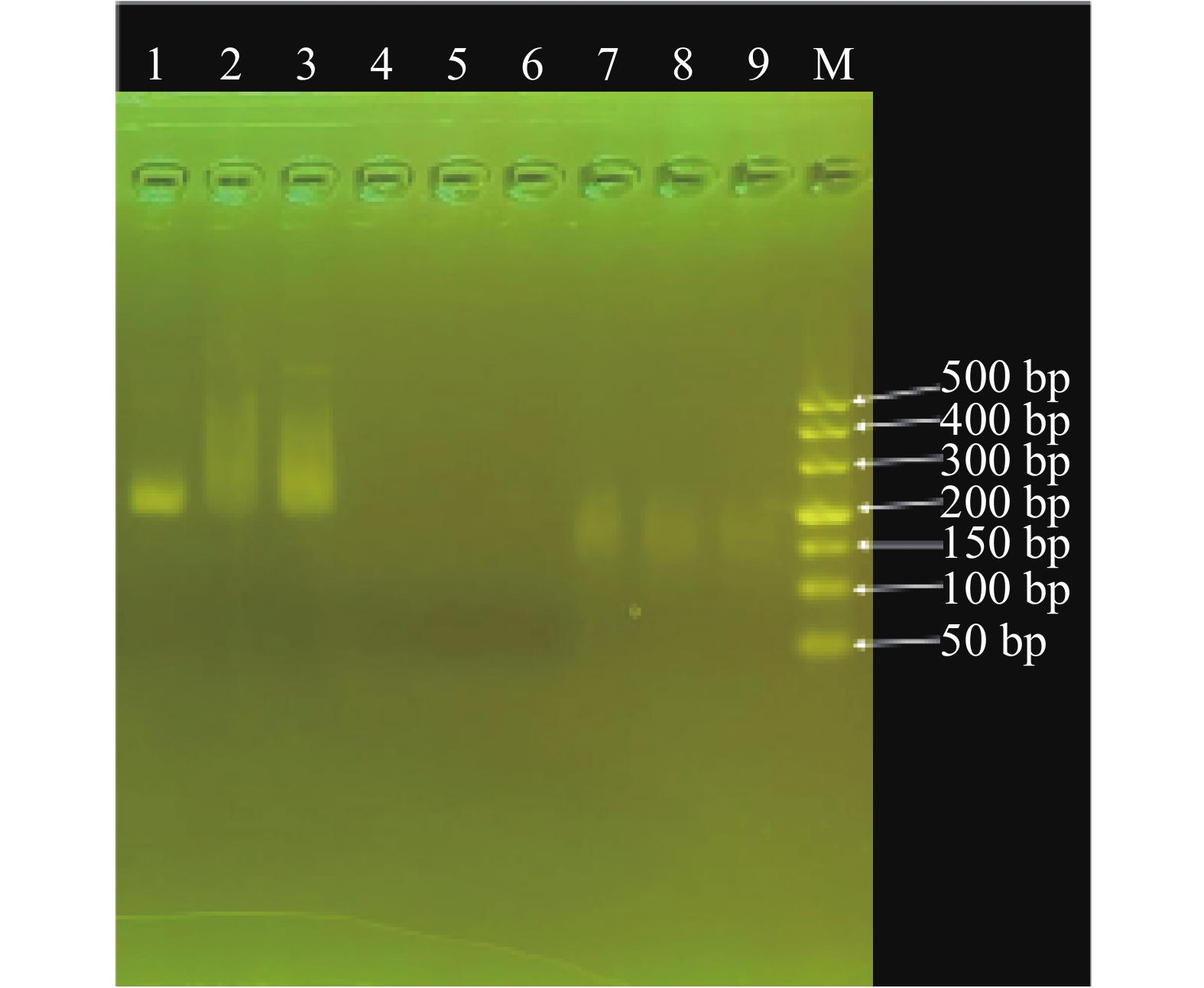
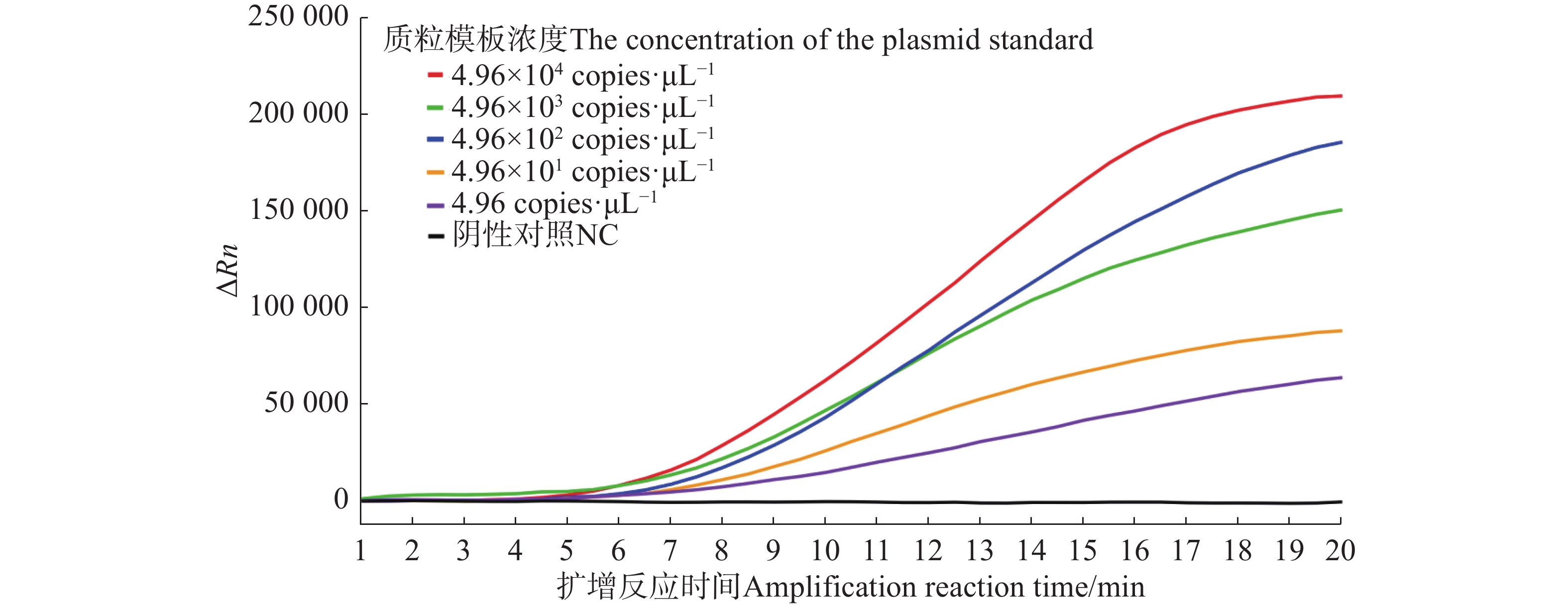
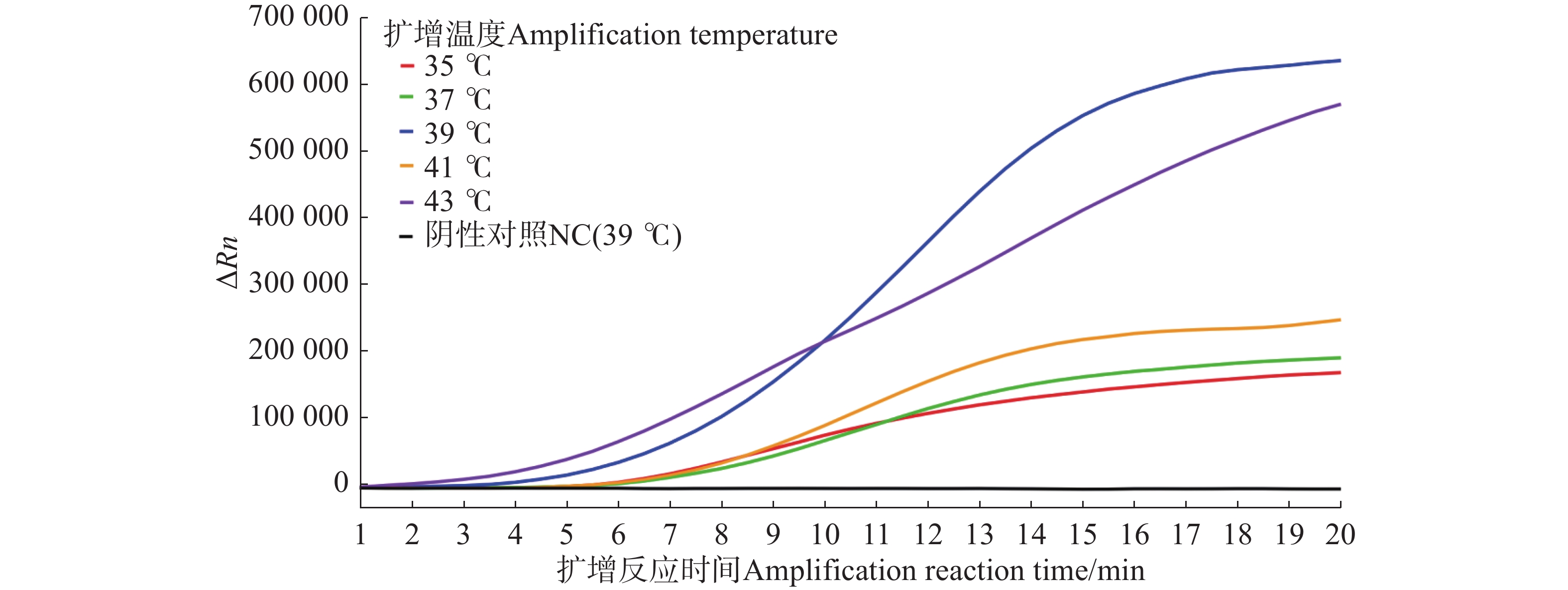
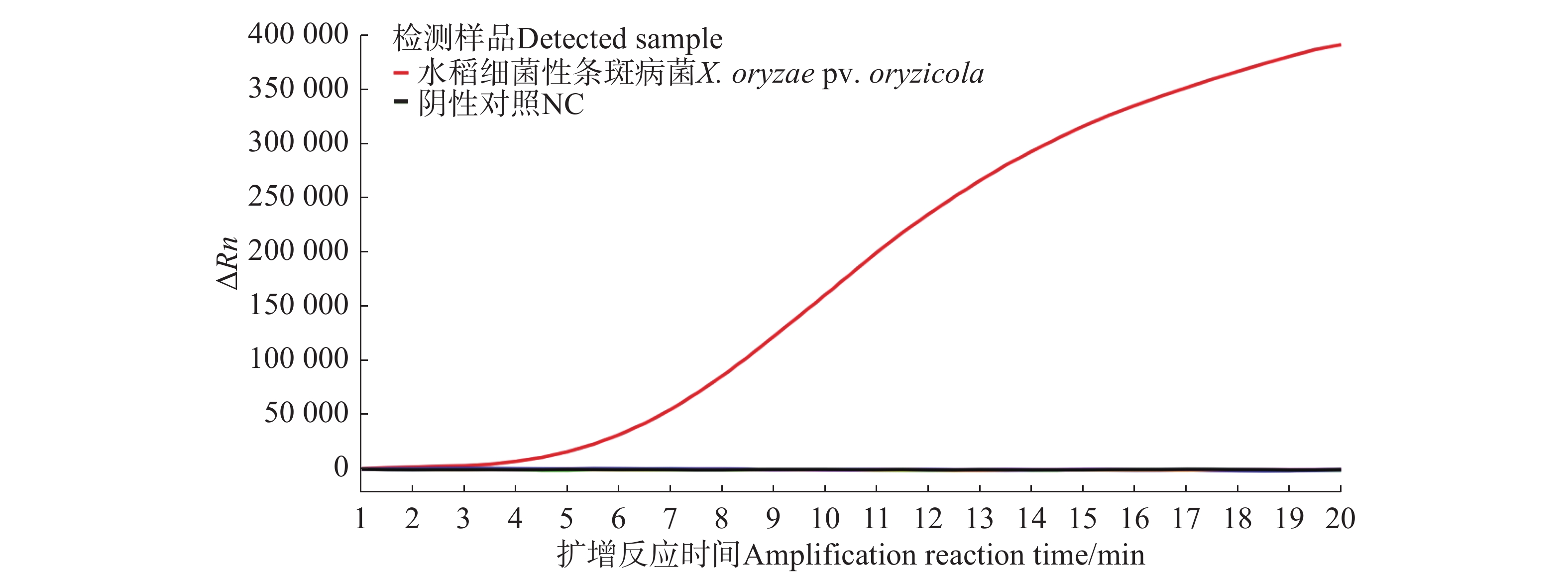
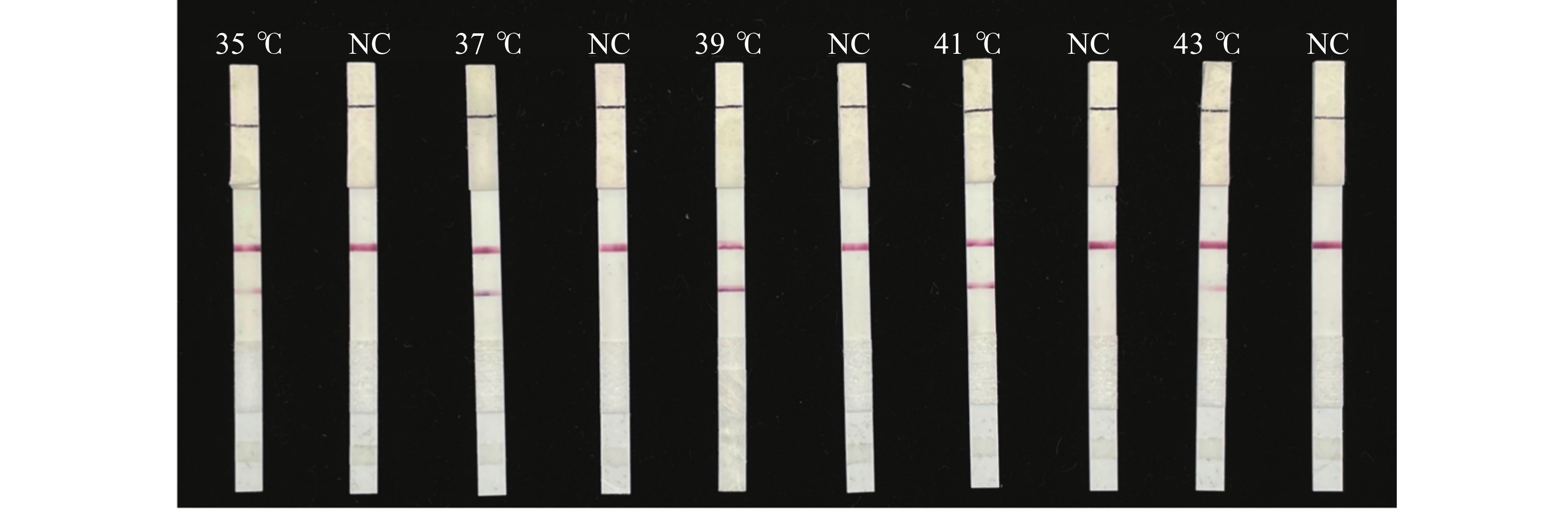
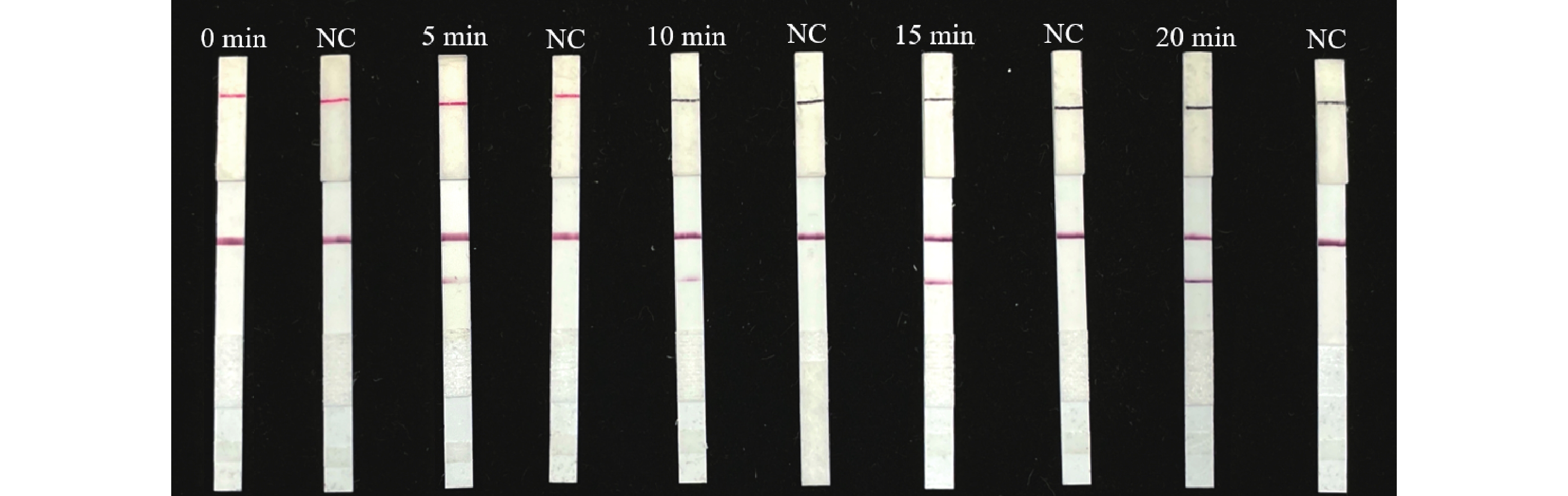
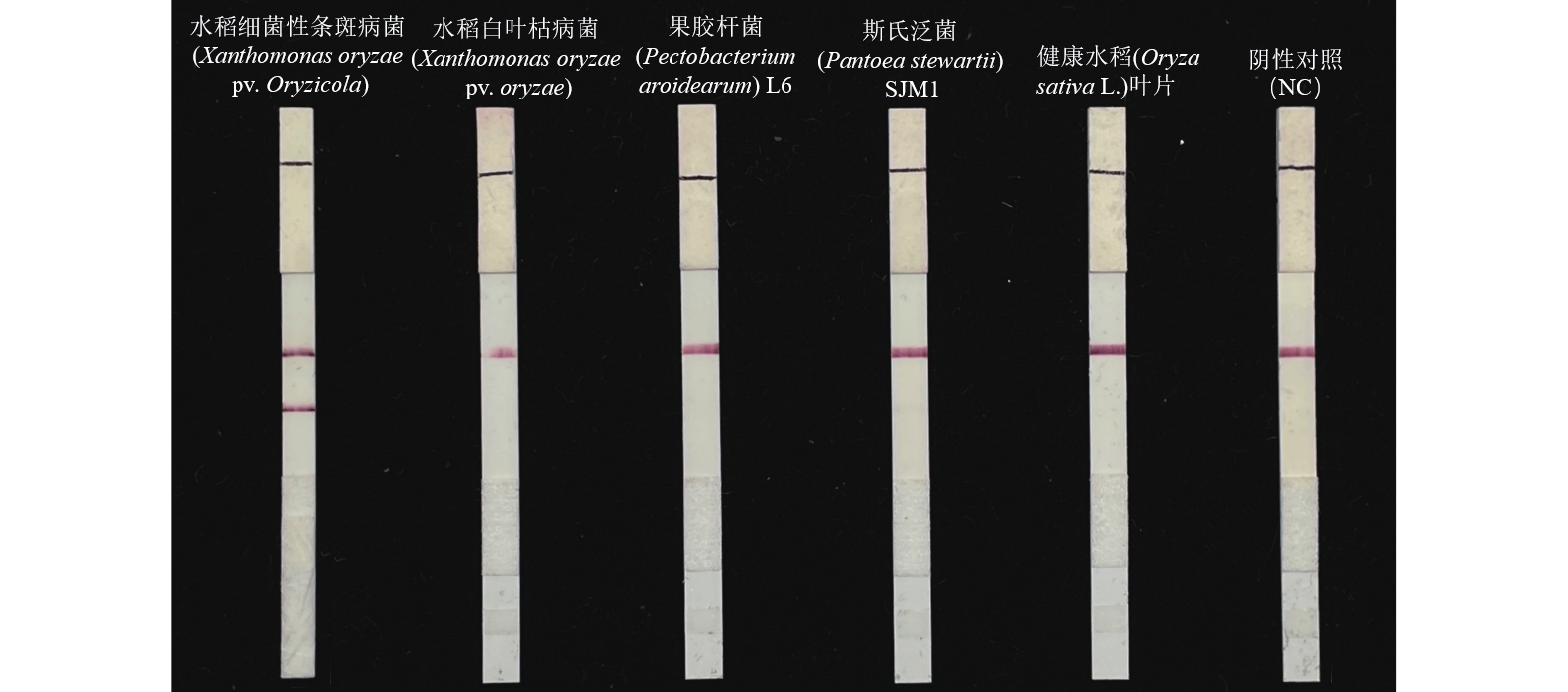
For early and rapid detection of Xanthomonas oryzae pv. oryzicola (Xoc), the cause of bacterial leaf streak in rice, specific recombinase polymerase amplification (RPA) primers, and Exo fluorescent and nfo probes were designed and screened, respectively, according to the publicly available gene sequence XOCgx_RS03090 of Xoc genome sequence in GenBank. The recombinant plasmid containing Xoc gene was constructed as a positive control. One fluorescent Exo-RPA assay and the other lateral flow strip detection (LFD-RPA) assay were established for rapid detection of Xoc. The sensitivity and specificity of the RPA methods were assessed, and the amplification effects of the RPA methods were explored by optimizing the reaction temperature and reaction time. The results showed that the sensitivity of both the Exo-RPA and LFD-RPA assays for the detection of Xoc were both 4.96 copies·μL−1, and that the optimal temperatures for the reaction were both 39 ℃. Xoc was detected within 12 min by the optimized Exo-RPA method, and within 5 min by the optimized LFD-RPA. The Exo-RPA and LFD-RPA assays developed were highly specific and could specifically and uniquely identify Xoc among plant pathogens in test. Both the RPA methods are highly sensitive, specific and fast, which pose a high potential to the monitoring of the target pathogen and early diagnosis of the disease.
For early and rapid detection of Xanthomonas oryzae pv. oryzicola (Xoc), the cause of bacterial leaf streak in rice, specific recombinase polymerase amplification (RPA) primers, and Exo fluorescent and nfo probes were designed and screened, respectively, according to the publicly available gene sequence XOCgx_RS03090 of Xoc genome sequence in GenBank. The recombinant plasmid containing Xoc gene was constructed as a positive control. One fluorescent Exo-RPA assay and the other lateral flow strip detection (LFD-RPA) assay were established for rapid detection of Xoc. The sensitivity and specificity of the RPA methods were assessed, and the amplification effects of the RPA methods were explored by optimizing the reaction temperature and reaction time. The results showed that the sensitivity of both the Exo-RPA and LFD-RPA assays for the detection of Xoc were both 4.96 copies·μL−1, and that the optimal temperatures for the reaction were both 39 ℃. Xoc was detected within 12 min by the optimized Exo-RPA method, and within 5 min by the optimized LFD-RPA. The Exo-RPA and LFD-RPA assays developed were highly specific and could specifically and uniquely identify Xoc among plant pathogens in test. Both the RPA methods are highly sensitive, specific and fast, which pose a high potential to the monitoring of the target pathogen and early diagnosis of the disease.

2026, 17(3): 508-522.
doi: 10.15886/j.cnki.rdswxb.20250030
Abstract:
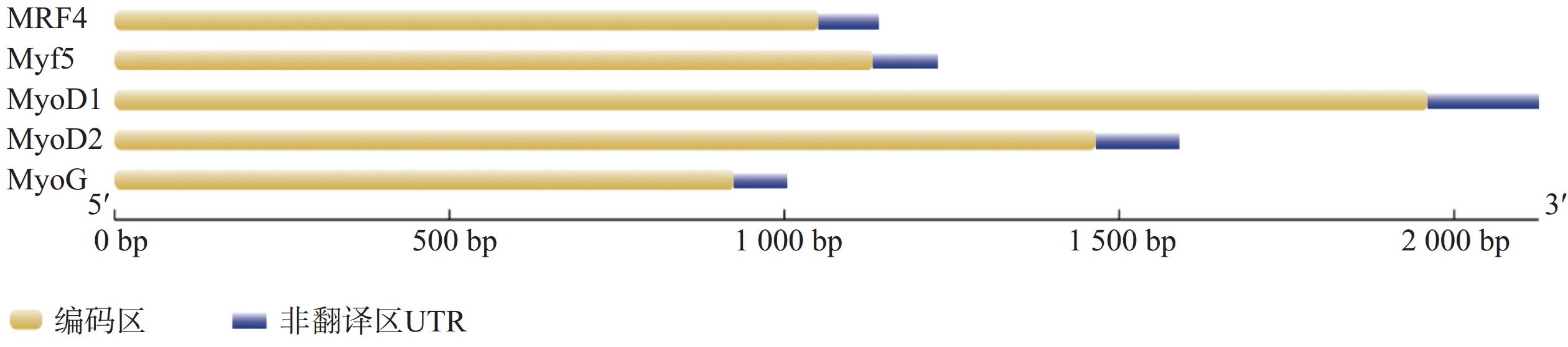
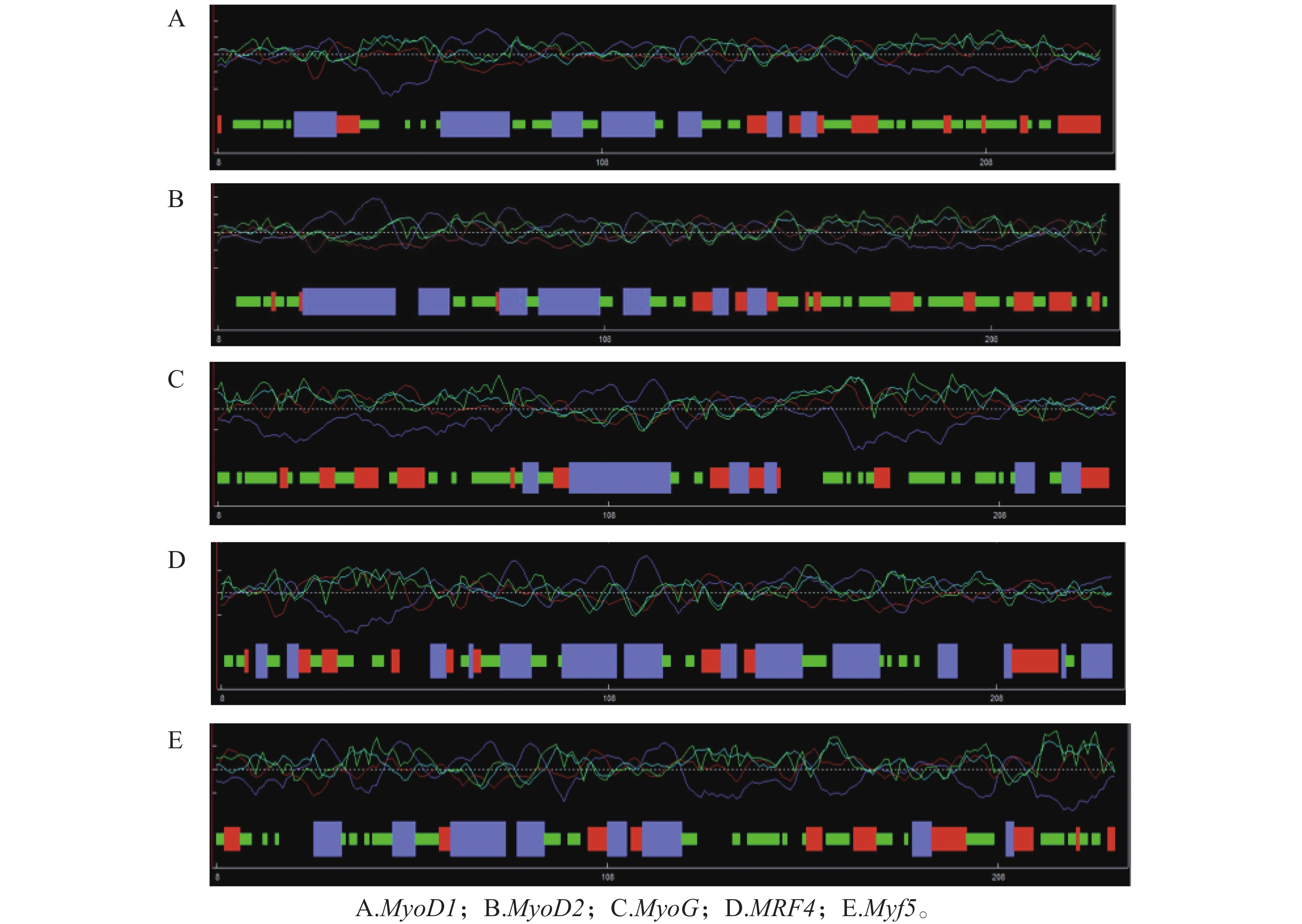
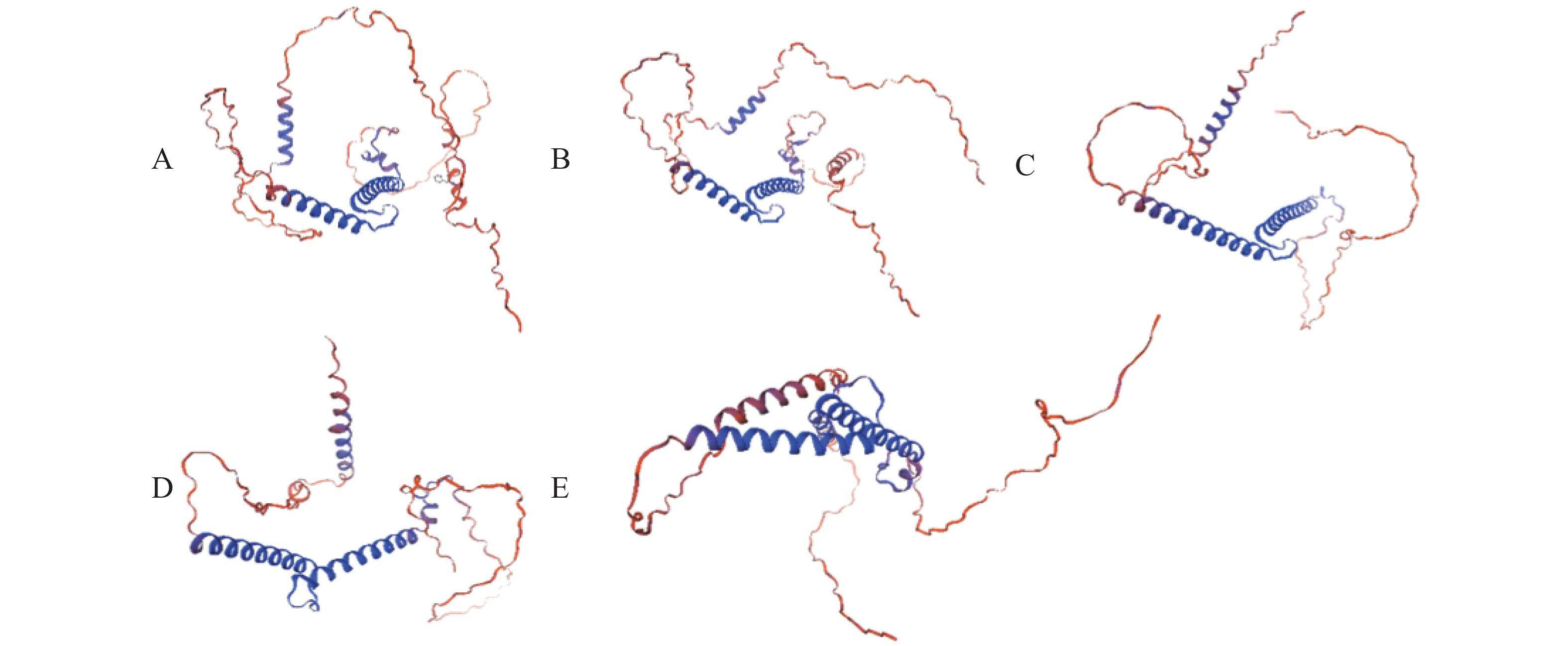
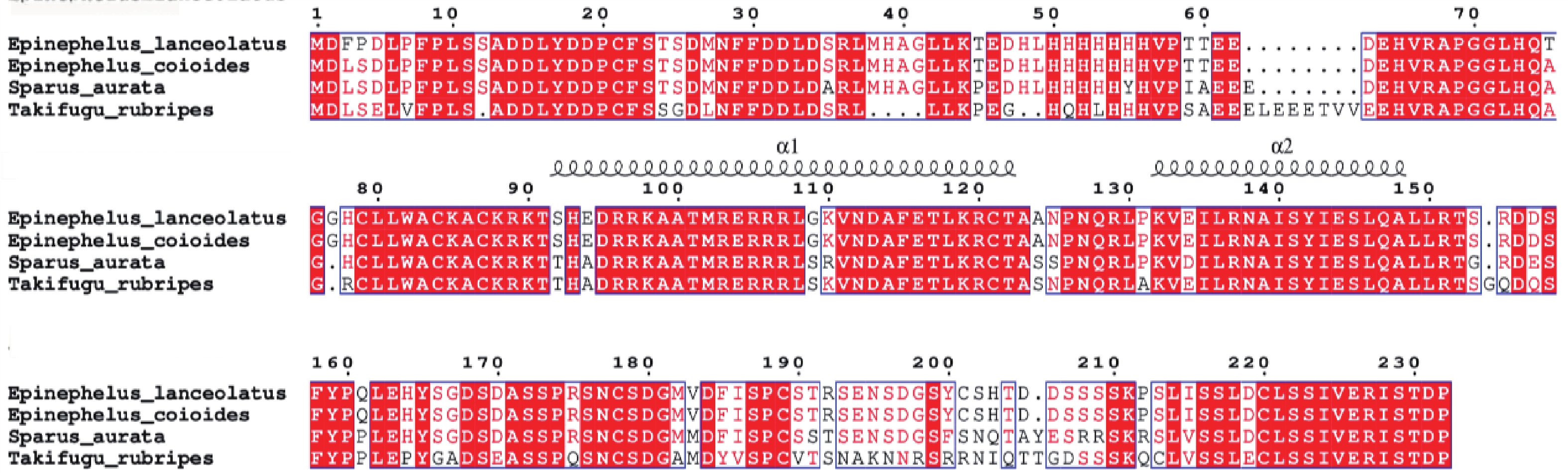
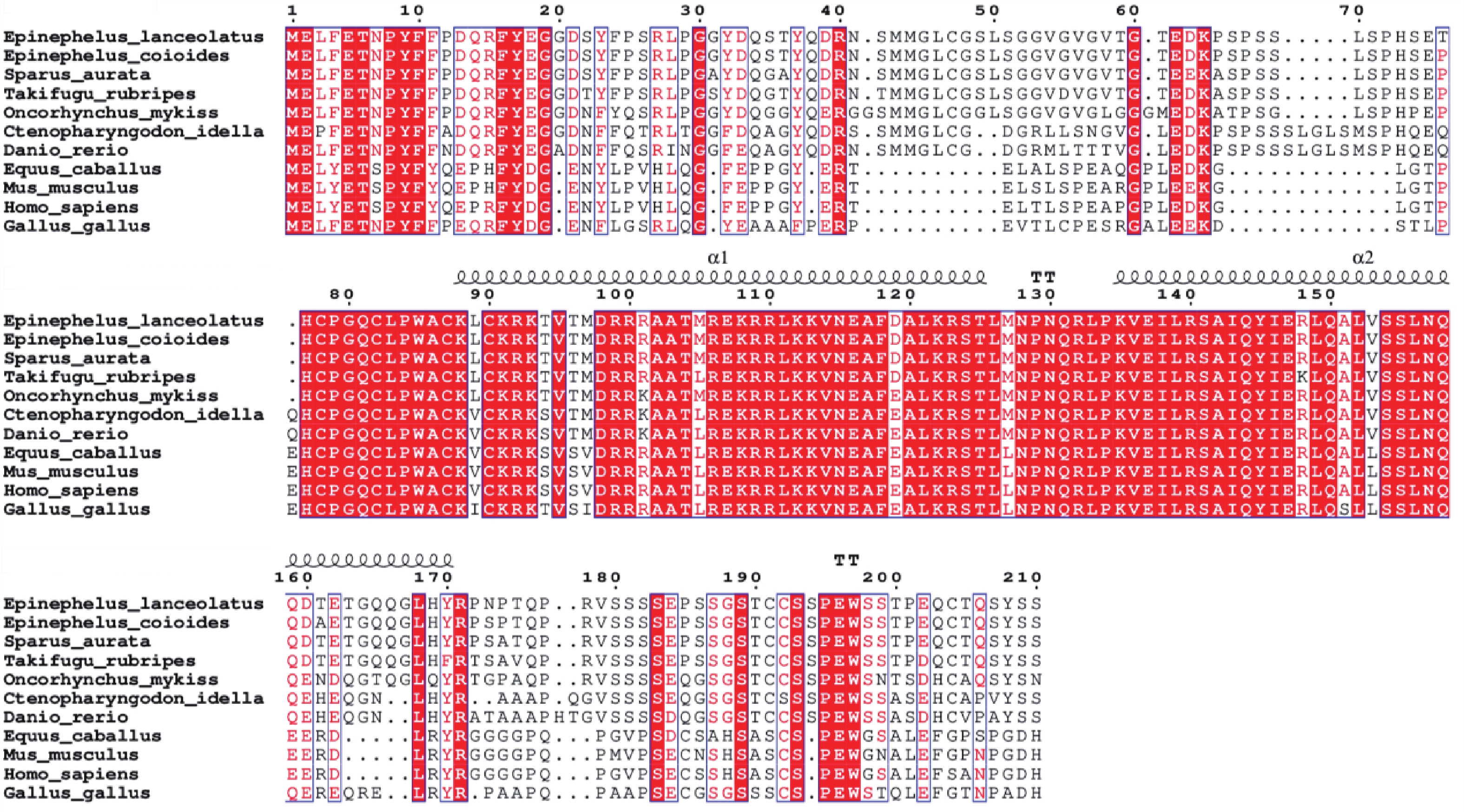
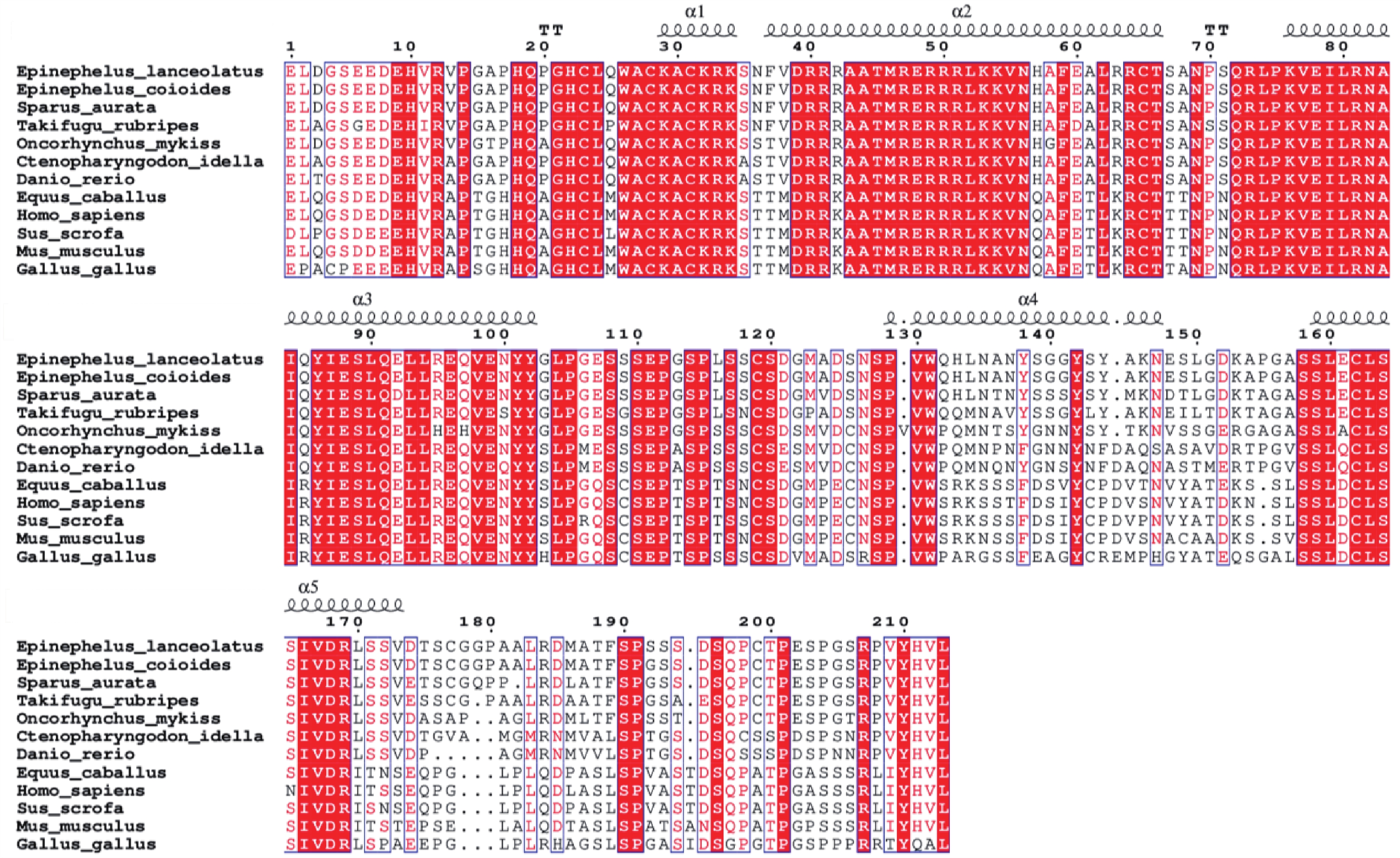
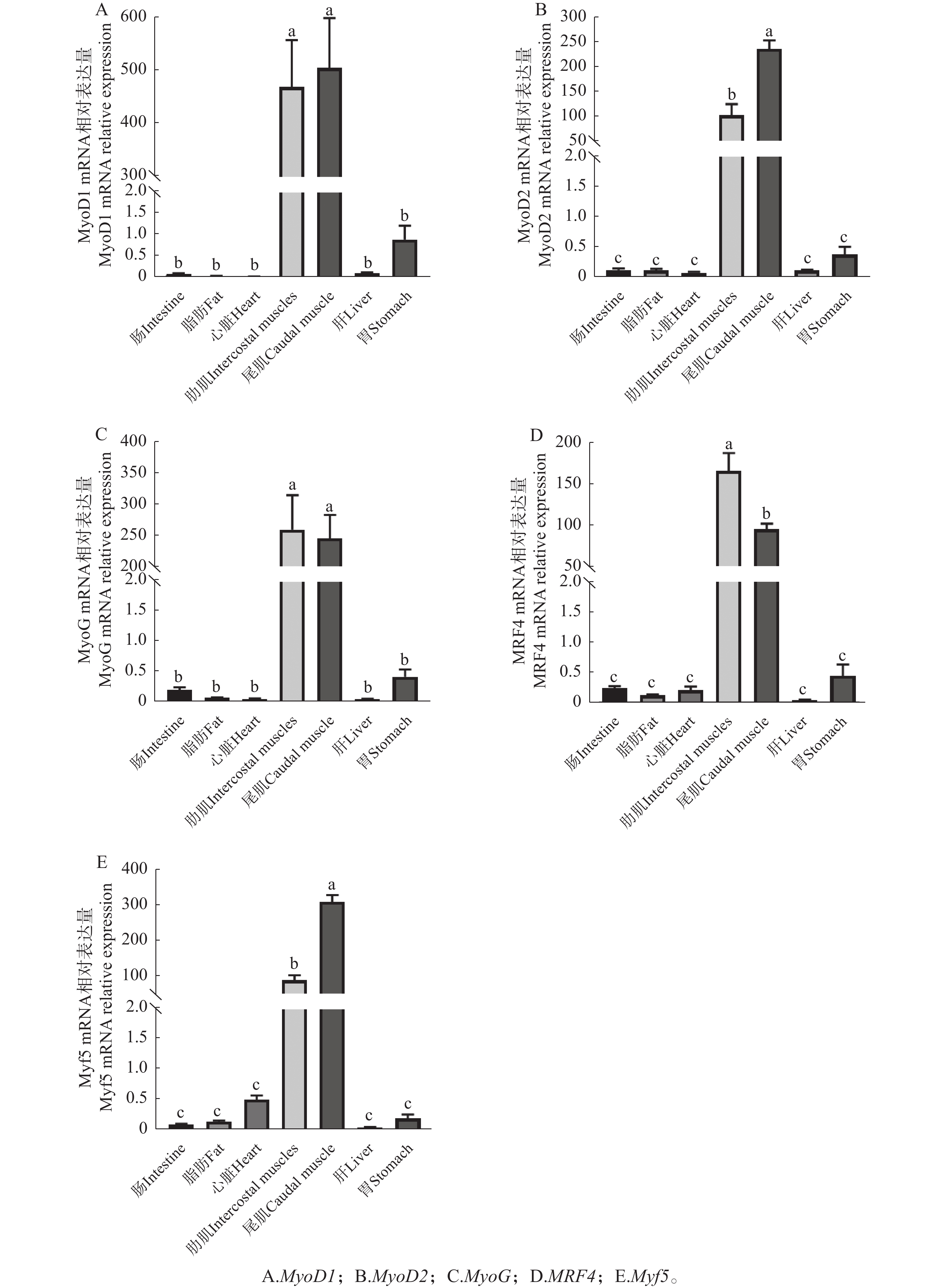
Myogenic regulatory factors (MRFs) are critical regulators of skeletal muscle growth and development in animals. The full-length cDNA sequences of five MRFs from the giant grouper (Epinephelus lanceolatus) were cloned using RT-PCR and RAC, including MyoD1 (1,962 bp), MyoD2 (1,466 bp), MyoG (926 bp), MRF4 (1,052 bp), and Myf5 (1,133 bp), encoding 297, 270, 251, 238, and 242 amino acids, respectively. Amino acid sequence alignment revealed that all five MRFs contain a conserved basic helix-loop-helix (bHLH) domain consisting of 60 amino acids. Phylogenetic analysis demonstrated that the MRF genes are clustered into two distinct branches: MyoD (MyoD1 and MyoD2) groups with Myf5 in one clade, while MRF4 and MyoG form the other. All genes showed closest evolutionary relationships with Perciformes homologs. Real-time quantitative PCR analysis revealed predominant expression of these MRFs in skeletal muscle, with significantly lower expression levels in the liver, heart, and intestine. These findings provide foundation for elucidating the regulatory mechanisms of MRFs in skeletal muscle development of the giant grouper.
Myogenic regulatory factors (MRFs) are critical regulators of skeletal muscle growth and development in animals. The full-length cDNA sequences of five MRFs from the giant grouper (Epinephelus lanceolatus) were cloned using RT-PCR and RAC, including MyoD1 (1,962 bp), MyoD2 (1,466 bp), MyoG (926 bp), MRF4 (1,052 bp), and Myf5 (1,133 bp), encoding 297, 270, 251, 238, and 242 amino acids, respectively. Amino acid sequence alignment revealed that all five MRFs contain a conserved basic helix-loop-helix (bHLH) domain consisting of 60 amino acids. Phylogenetic analysis demonstrated that the MRF genes are clustered into two distinct branches: MyoD (MyoD1 and MyoD2) groups with Myf5 in one clade, while MRF4 and MyoG form the other. All genes showed closest evolutionary relationships with Perciformes homologs. Real-time quantitative PCR analysis revealed predominant expression of these MRFs in skeletal muscle, with significantly lower expression levels in the liver, heart, and intestine. These findings provide foundation for elucidating the regulatory mechanisms of MRFs in skeletal muscle development of the giant grouper.




2026, 17(3): 523-531.
doi: 10.15886/j.cnki.rdswxb.20250077
Abstract:
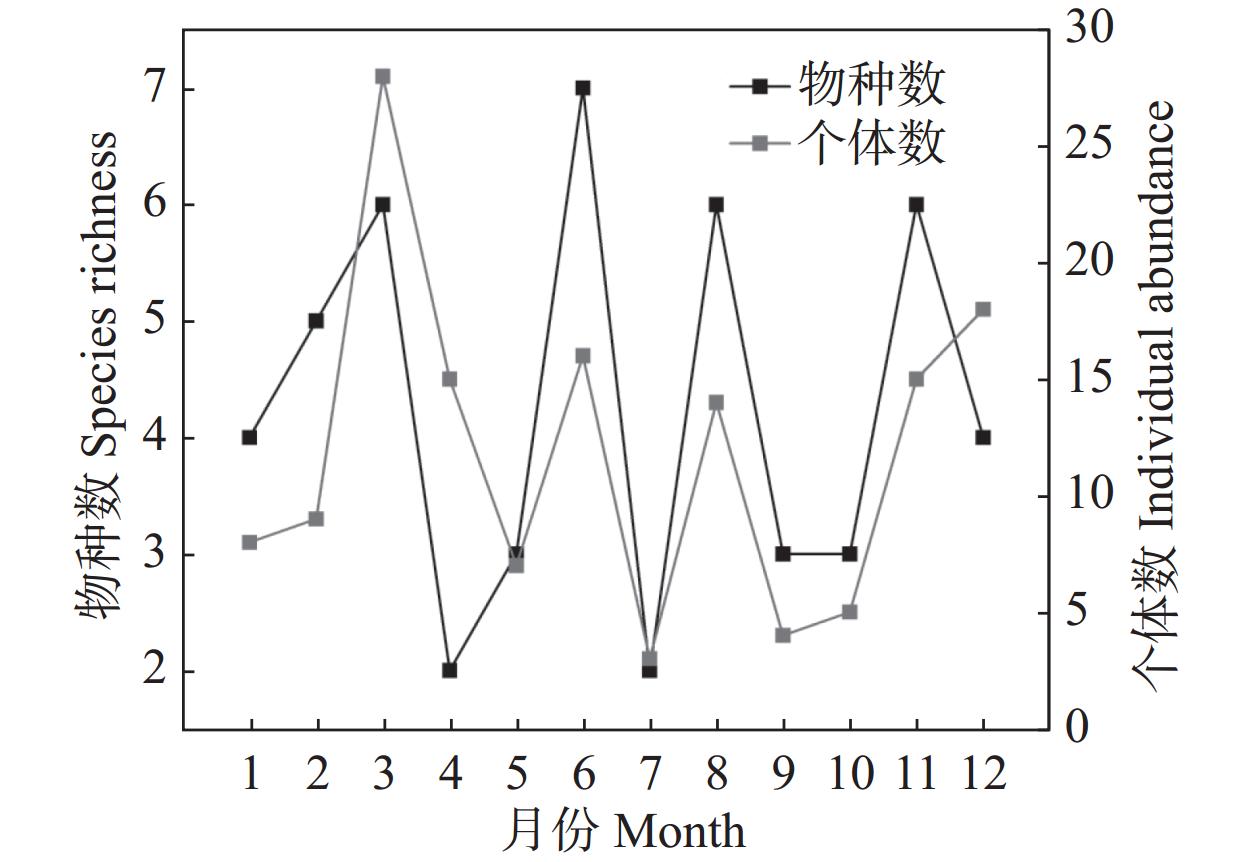
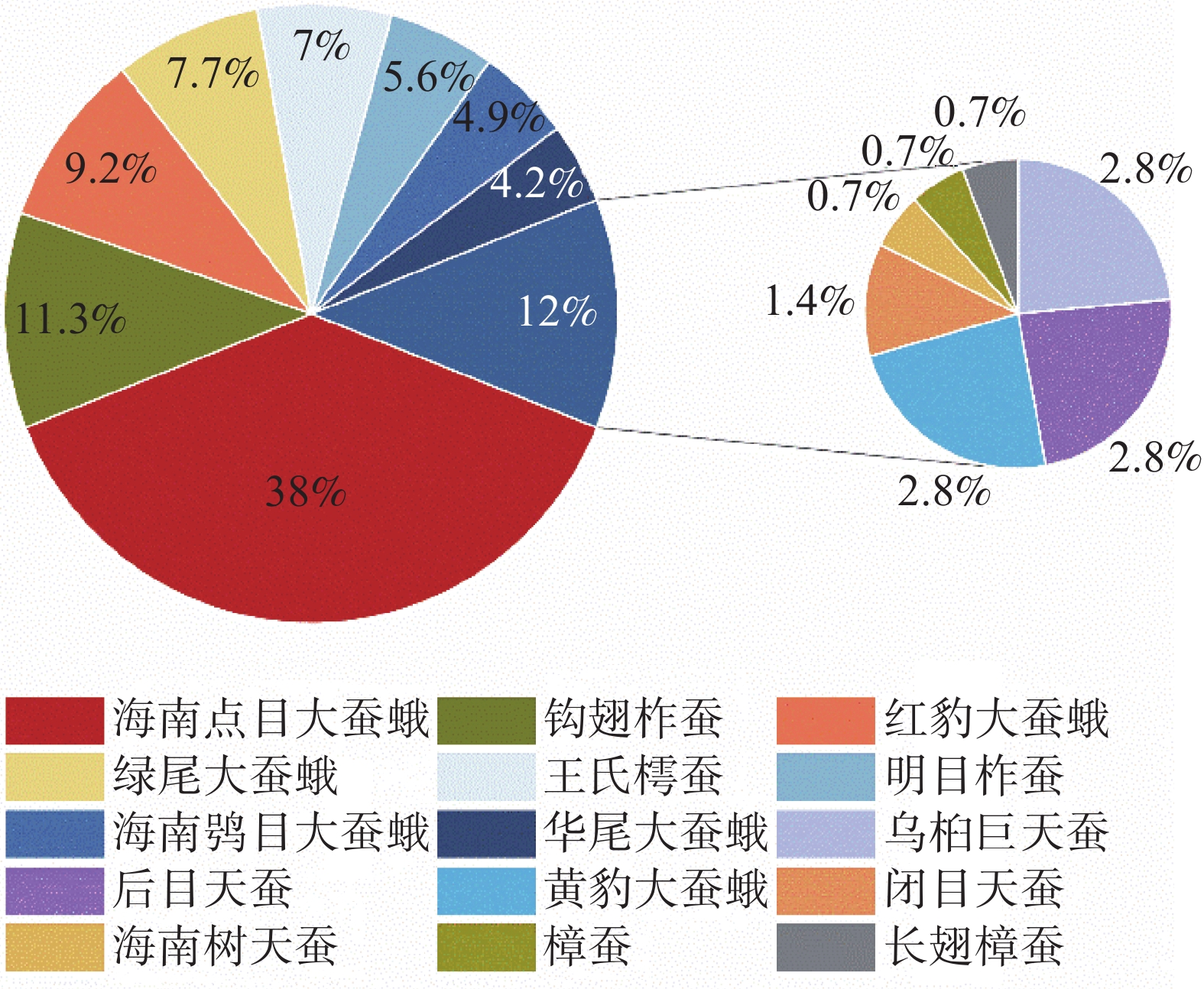
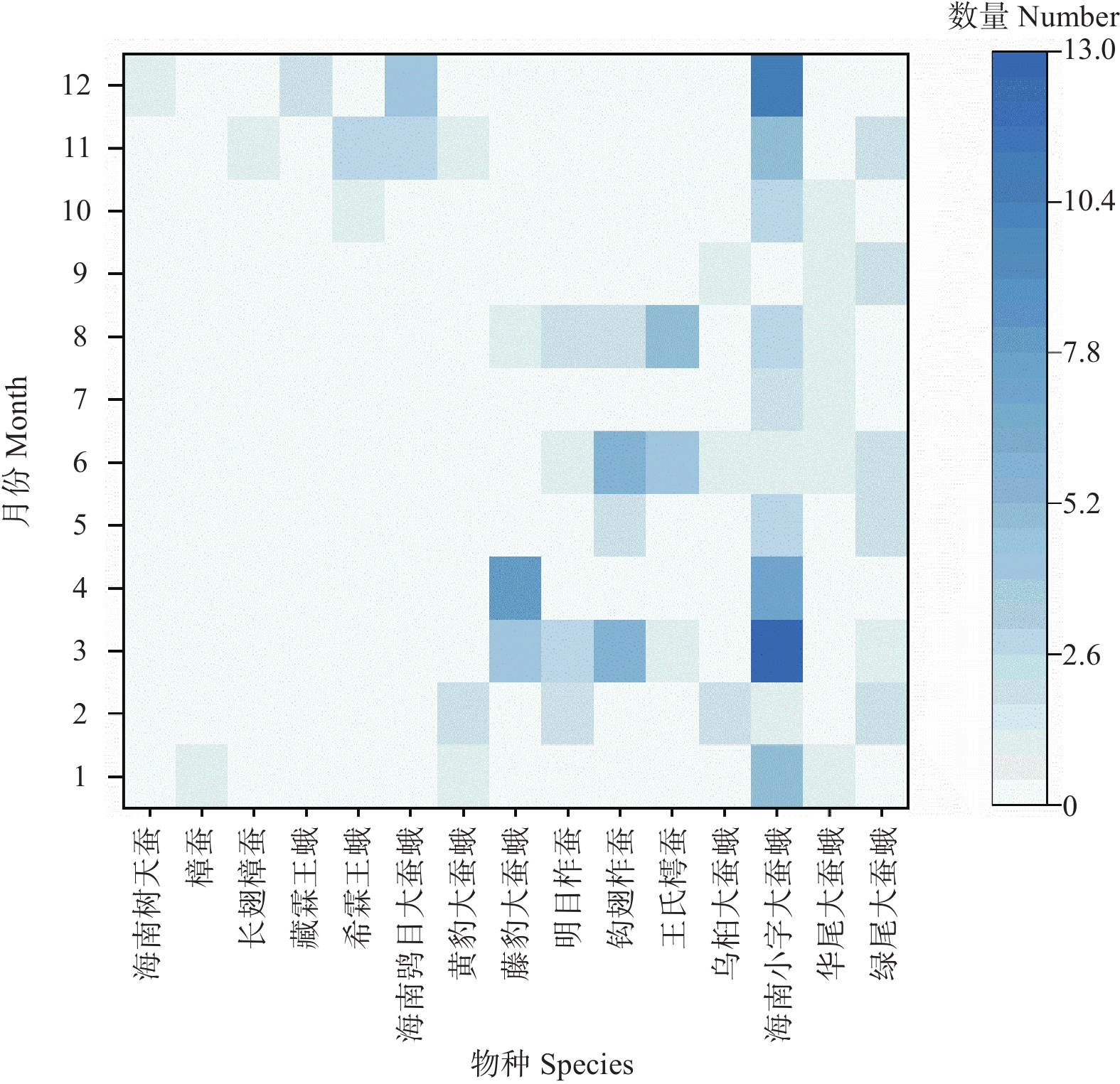
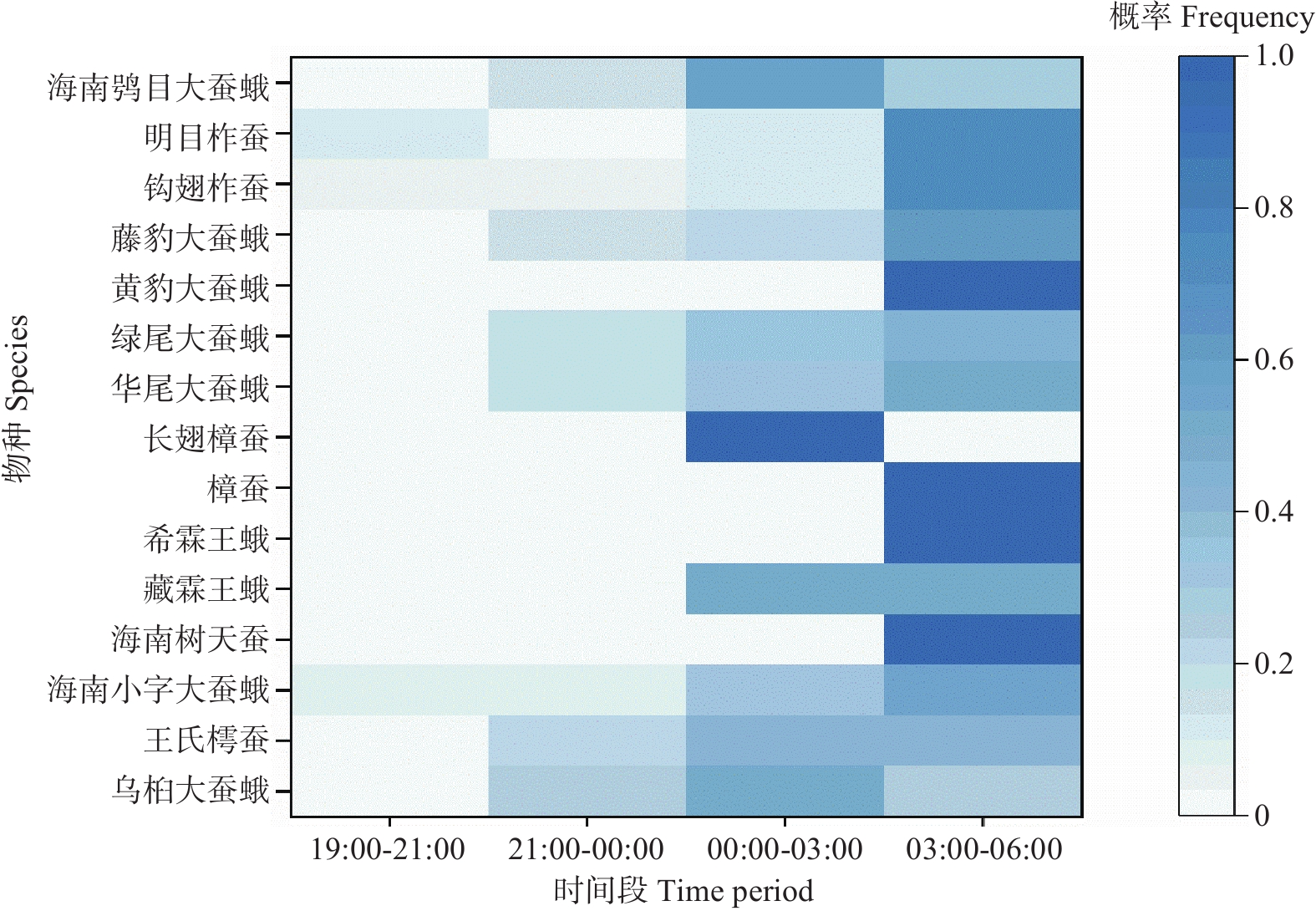
This study investigated the diversity and phenology of Saturniidae moths in Limushan, Hainan Tropical Rainforest National Park. Through light trapping and transect surveys from 2020 to 2024, we recorded 15 Saturniidae species comprising 2 subfamilies and 9 genera, including 3 Hainan-endemic species. Analysis of flight periods revealed peak activity between November and the following August of the following year, with 56.34% of moths showing peak phototactic response at 03:00–06:00. Laboratory rearing indicated 6 univoltine species, 2 bivoltine species, and 7 multivoltine species. The faunal composition of Saturniidae in Limushan aligned with tropical zoogeographic regions, reflecting the area's distinctive ecological environment and biodiversity. These findings offer critical data for future species conservation and resource utilization.
This study investigated the diversity and phenology of Saturniidae moths in Limushan, Hainan Tropical Rainforest National Park. Through light trapping and transect surveys from 2020 to 2024, we recorded 15 Saturniidae species comprising 2 subfamilies and 9 genera, including 3 Hainan-endemic species. Analysis of flight periods revealed peak activity between November and the following August of the following year, with 56.34% of moths showing peak phototactic response at 03:00–06:00. Laboratory rearing indicated 6 univoltine species, 2 bivoltine species, and 7 multivoltine species. The faunal composition of Saturniidae in Limushan aligned with tropical zoogeographic regions, reflecting the area's distinctive ecological environment and biodiversity. These findings offer critical data for future species conservation and resource utilization.
2026, 17(3): 532-540.
doi: 10.15886/j.cnki.rdswxb.20240197
Abstract:
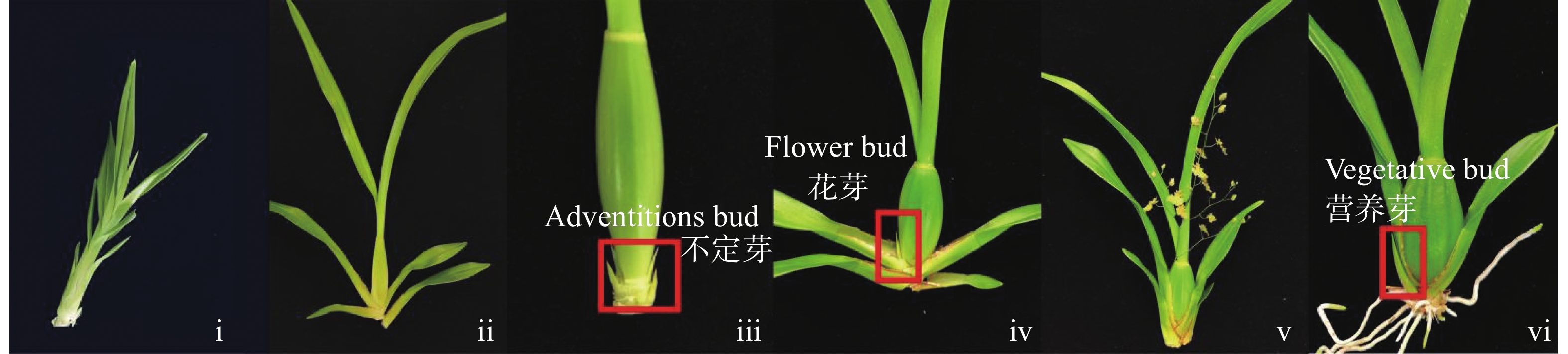
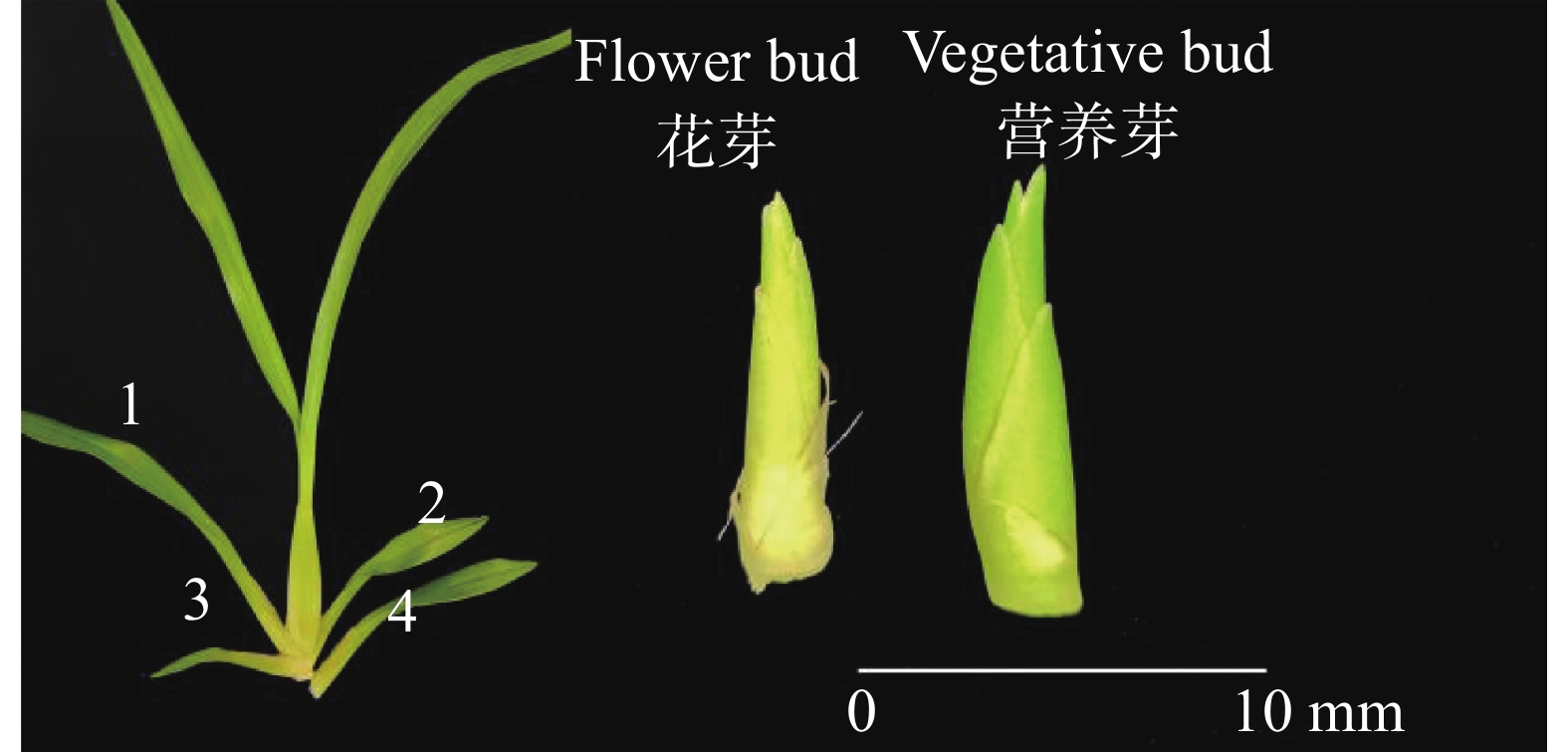
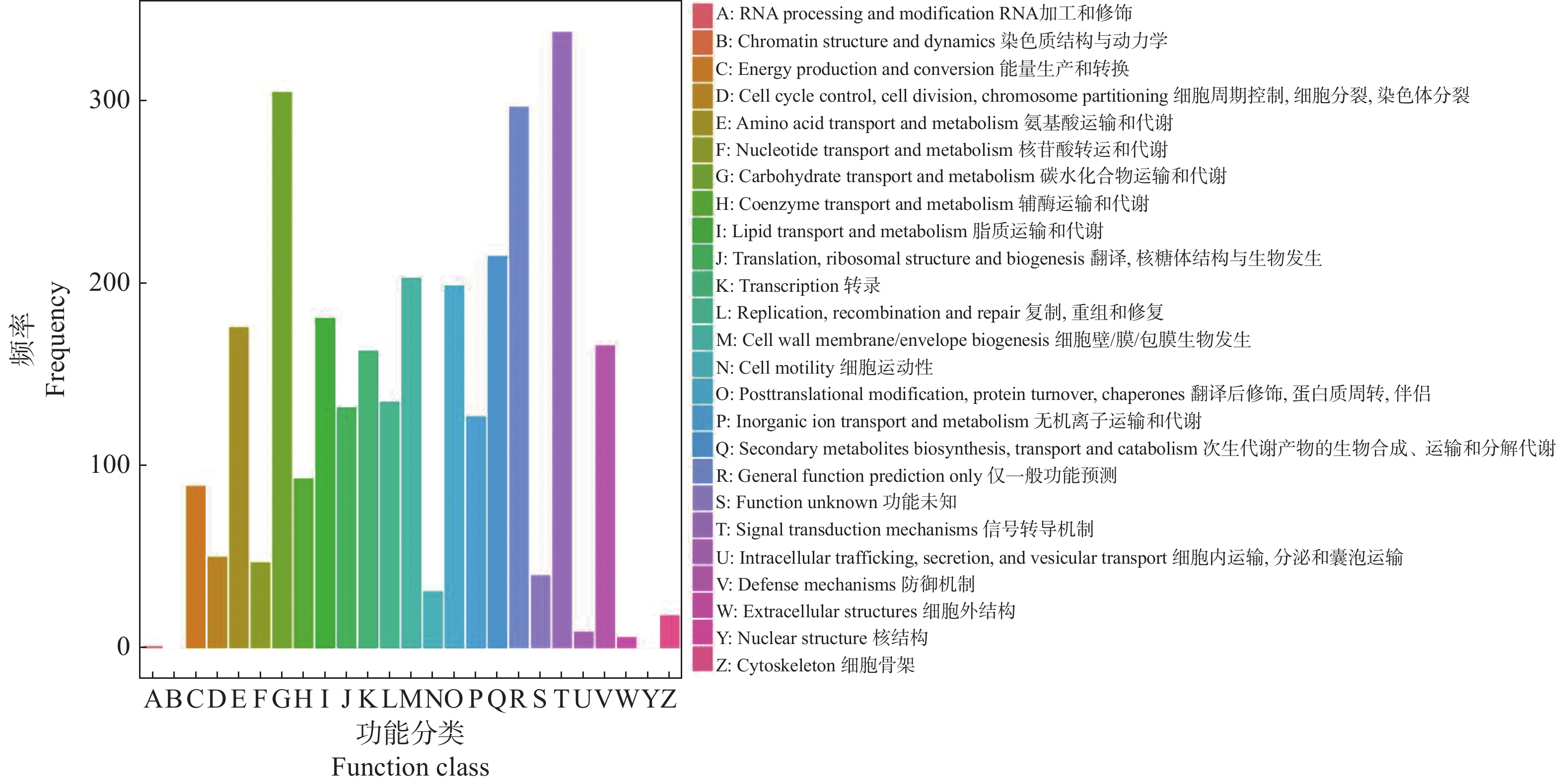
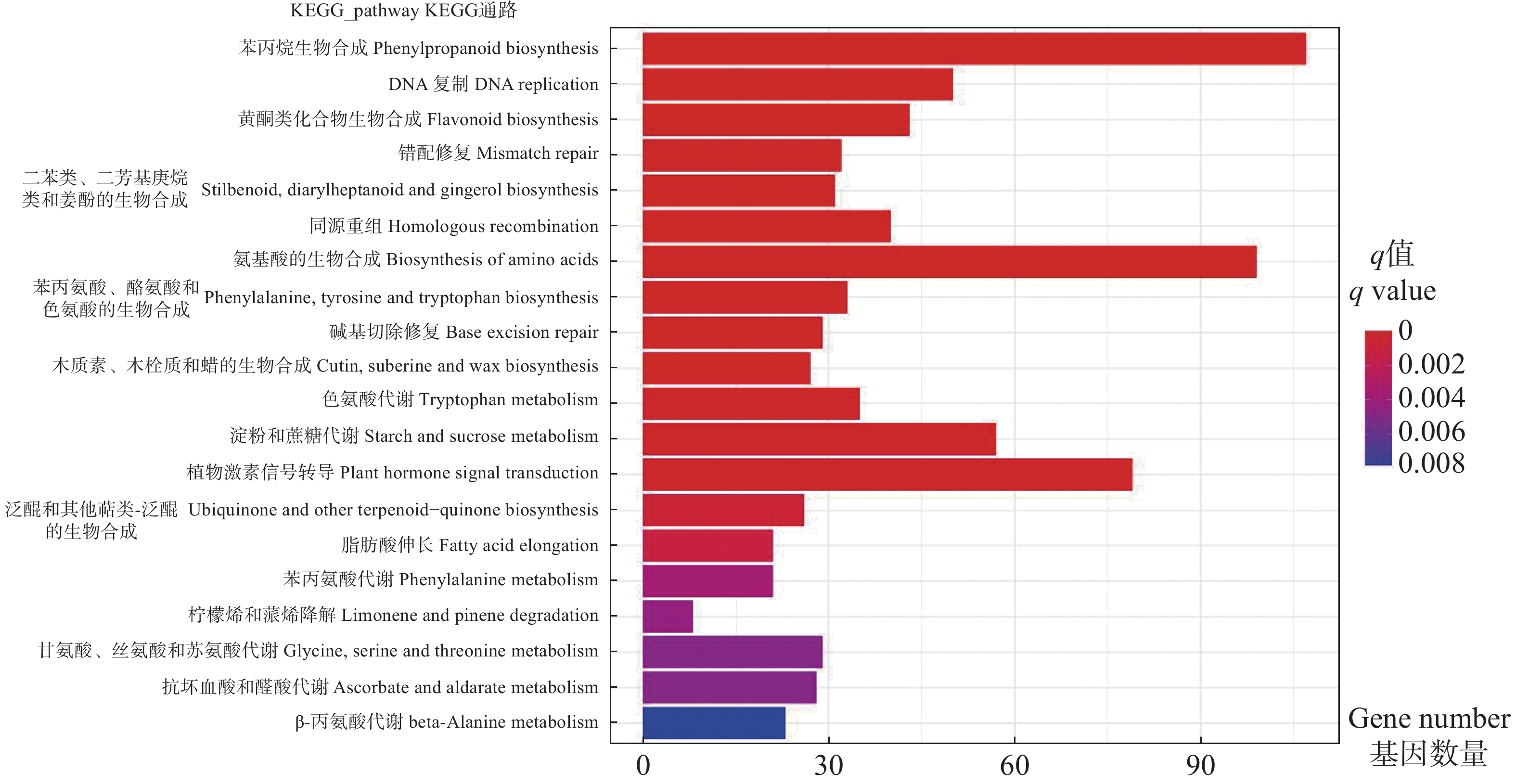
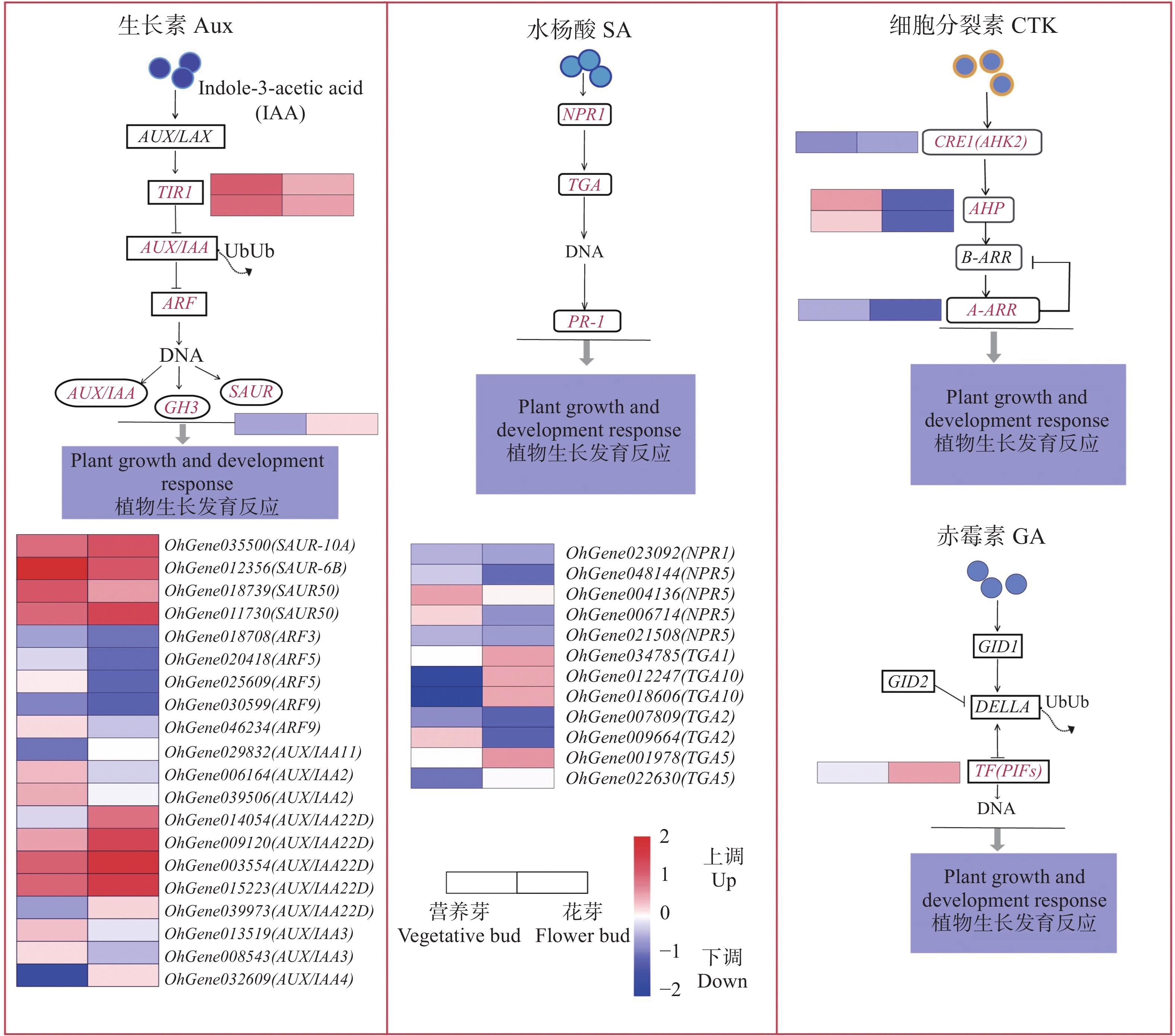
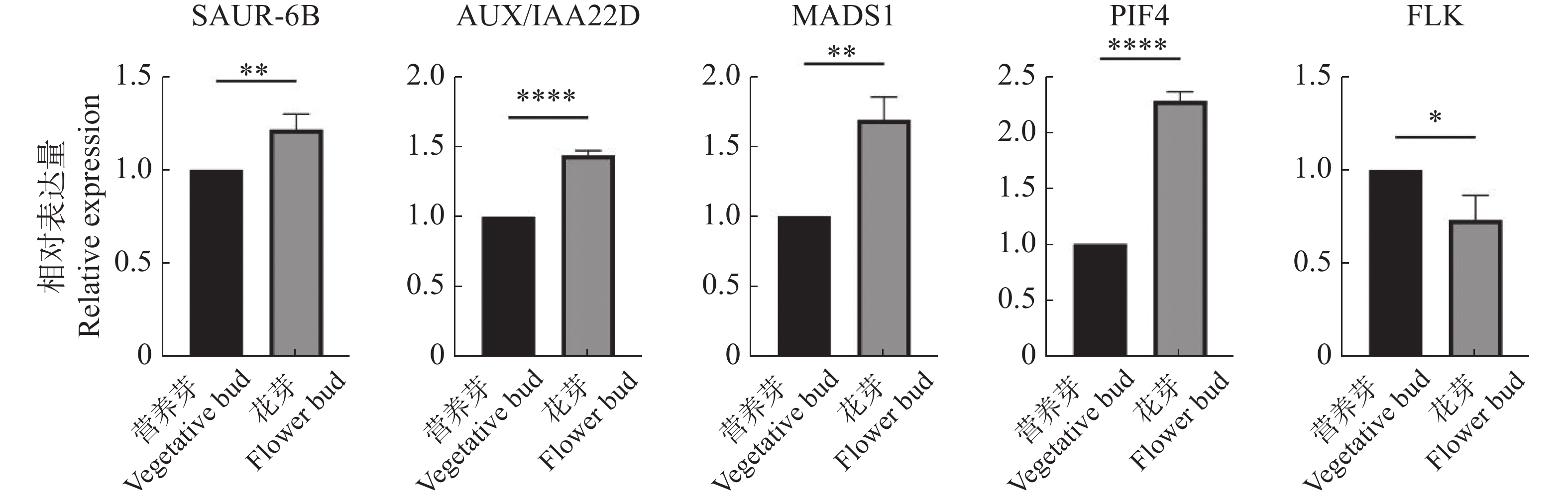
To address the decreased cut flower yield caused by the bud jumping development phenomenon, where adventitious buds interrupt the normal growth cycle of flower bud differentiation in Oncidium cut flower production, flower buds and vegetative buds of Oncidium hybridum 'Boda NO1' were selected for high-throughput transcriptome sequencing. A total of 127 452 717 high-quality sequences (37.36 Gb) were obtained, and 7 671 differentially expressed genes (DEGs) were identified. COG functional classification revealed primary enrichment in signal transduction pathways and carbohydrate transport/metabolism pathways. KEGG analysis showed that DEGs were significantly enriched in starch and sucrose metabolism, phytohormone signaling, and other pathways. Among these, 78 DEGs were identified in the phytohormone signaling pathway, with the most pronounced differences involving auxin, cytokinins, salicylic acid, and gibberellins. Thirteen transcription factors and flowering-related genes, including MADS1, AP2, and FLK, were also screened. These results partially elucidate the effects of auxin, cytokinins, and flowering-related genes on the differentiation of flower buds and vegetative buds in Oncidium. This study provides a theoretical foundation for further research on the mechanism underlying the bud jumping development phenomenon during Oncidium production. Additionally, it supports subsequent improvement in production efficiency, quality enhancement, and optimization of cultivation and management. These findings hold significant practical value for advancing orchid breeding, seedling production, and industry development in China.
To address the decreased cut flower yield caused by the bud jumping development phenomenon, where adventitious buds interrupt the normal growth cycle of flower bud differentiation in Oncidium cut flower production, flower buds and vegetative buds of Oncidium hybridum 'Boda NO1' were selected for high-throughput transcriptome sequencing. A total of 127 452 717 high-quality sequences (37.36 Gb) were obtained, and 7 671 differentially expressed genes (DEGs) were identified. COG functional classification revealed primary enrichment in signal transduction pathways and carbohydrate transport/metabolism pathways. KEGG analysis showed that DEGs were significantly enriched in starch and sucrose metabolism, phytohormone signaling, and other pathways. Among these, 78 DEGs were identified in the phytohormone signaling pathway, with the most pronounced differences involving auxin, cytokinins, salicylic acid, and gibberellins. Thirteen transcription factors and flowering-related genes, including MADS1, AP2, and FLK, were also screened. These results partially elucidate the effects of auxin, cytokinins, and flowering-related genes on the differentiation of flower buds and vegetative buds in Oncidium. This study provides a theoretical foundation for further research on the mechanism underlying the bud jumping development phenomenon during Oncidium production. Additionally, it supports subsequent improvement in production efficiency, quality enhancement, and optimization of cultivation and management. These findings hold significant practical value for advancing orchid breeding, seedling production, and industry development in China.
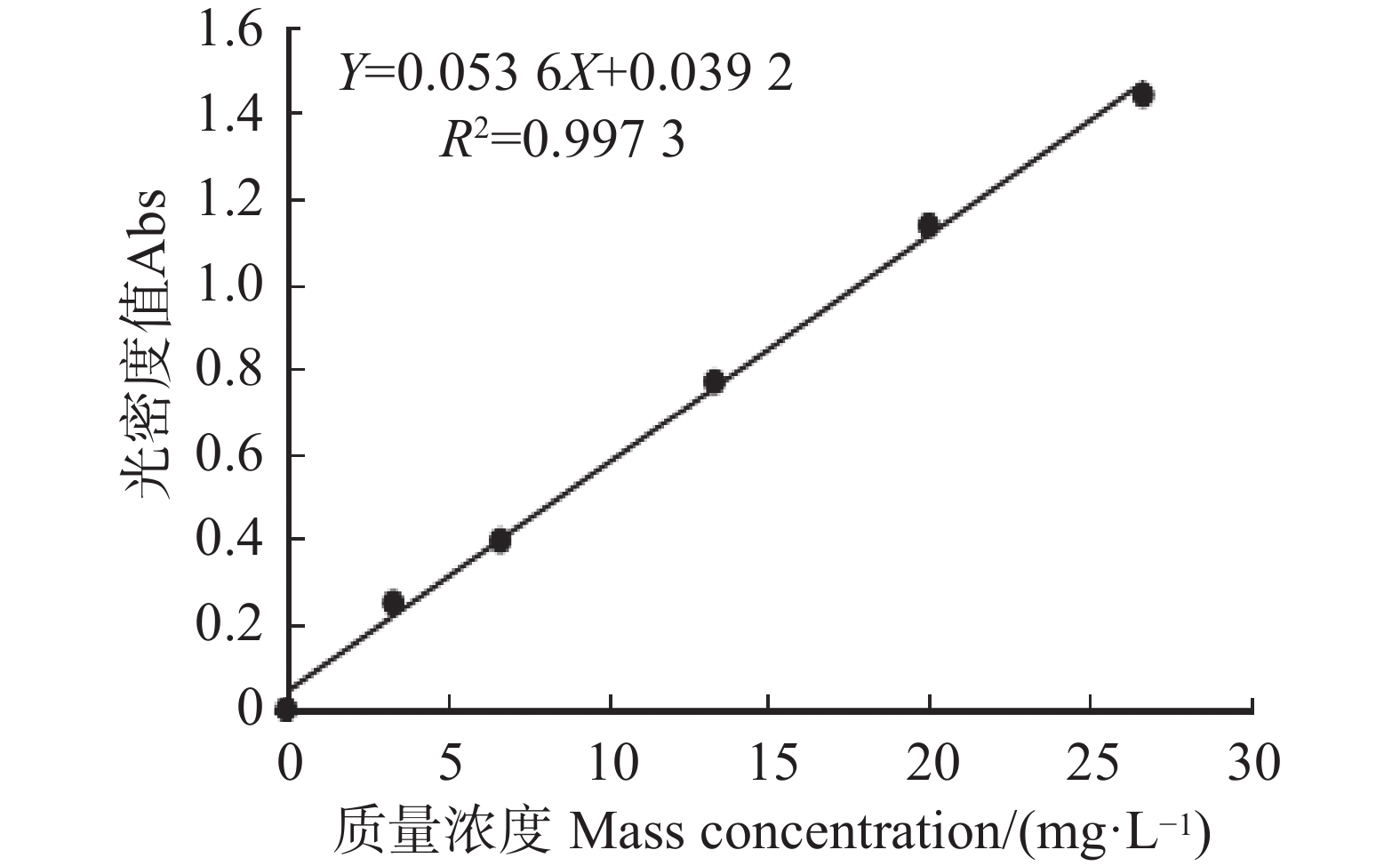
2026, 17(3): 541-548.
doi: 10.15886/j.cnki.rdswxb.20240088
Abstract:
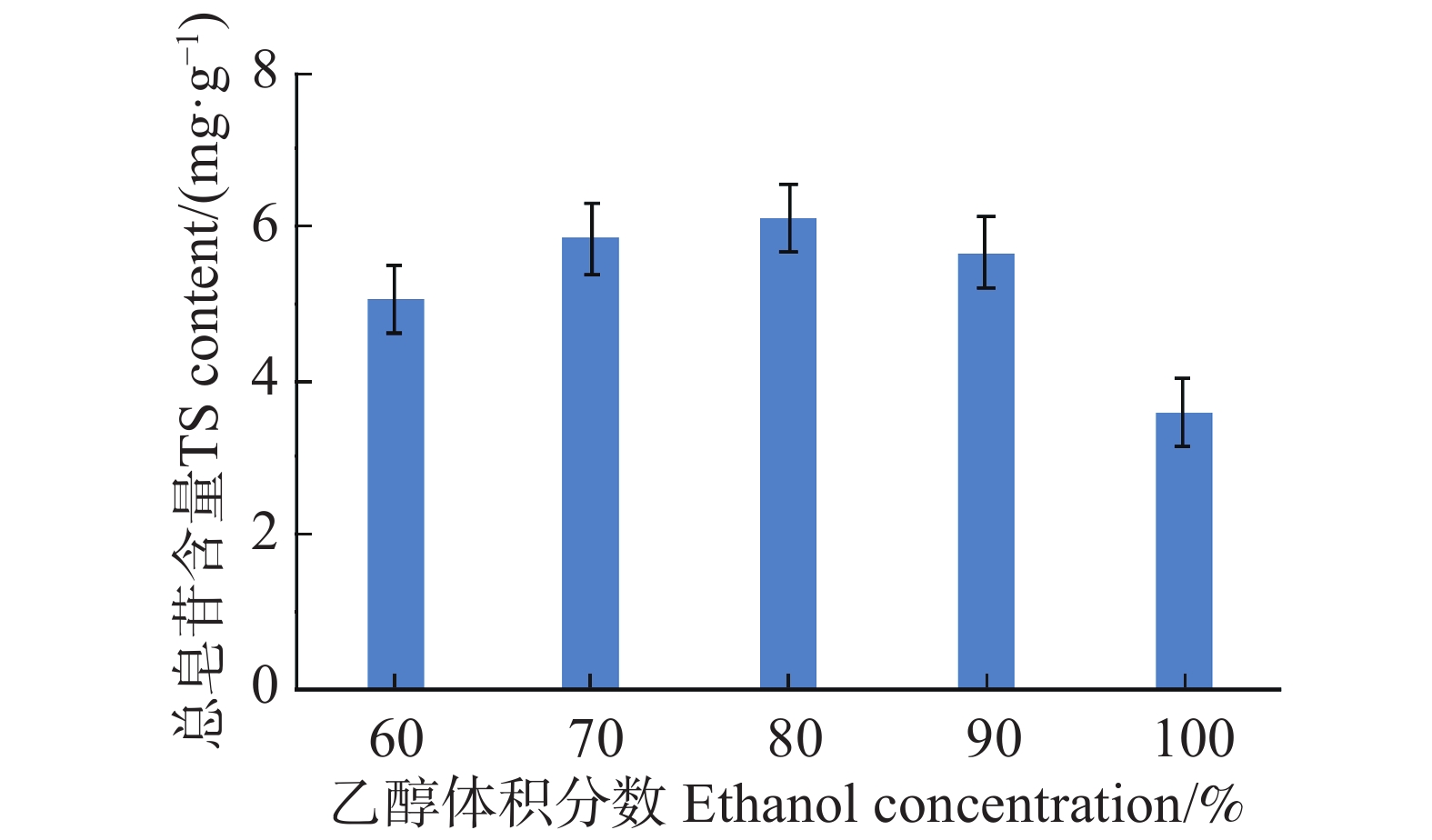
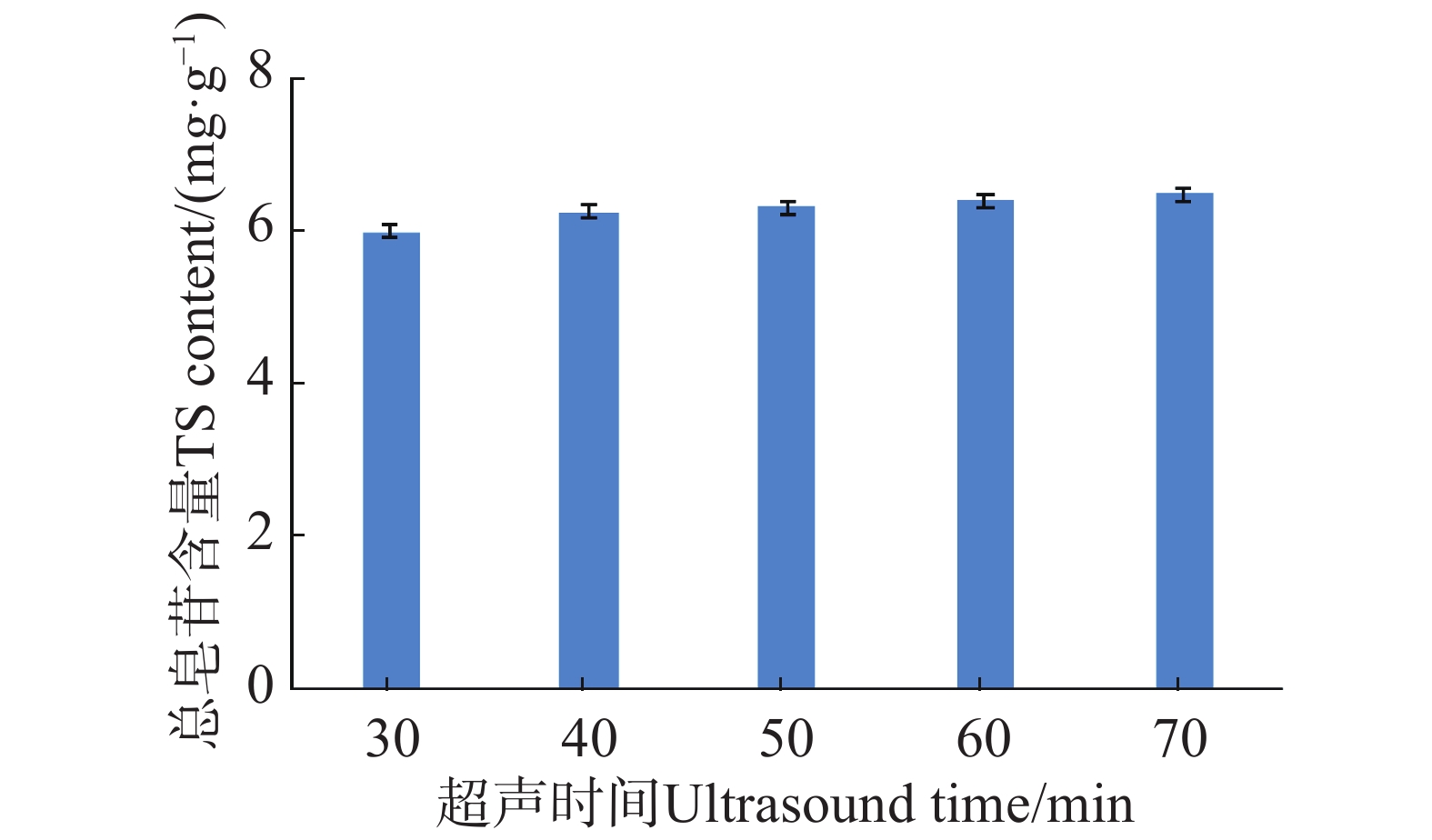
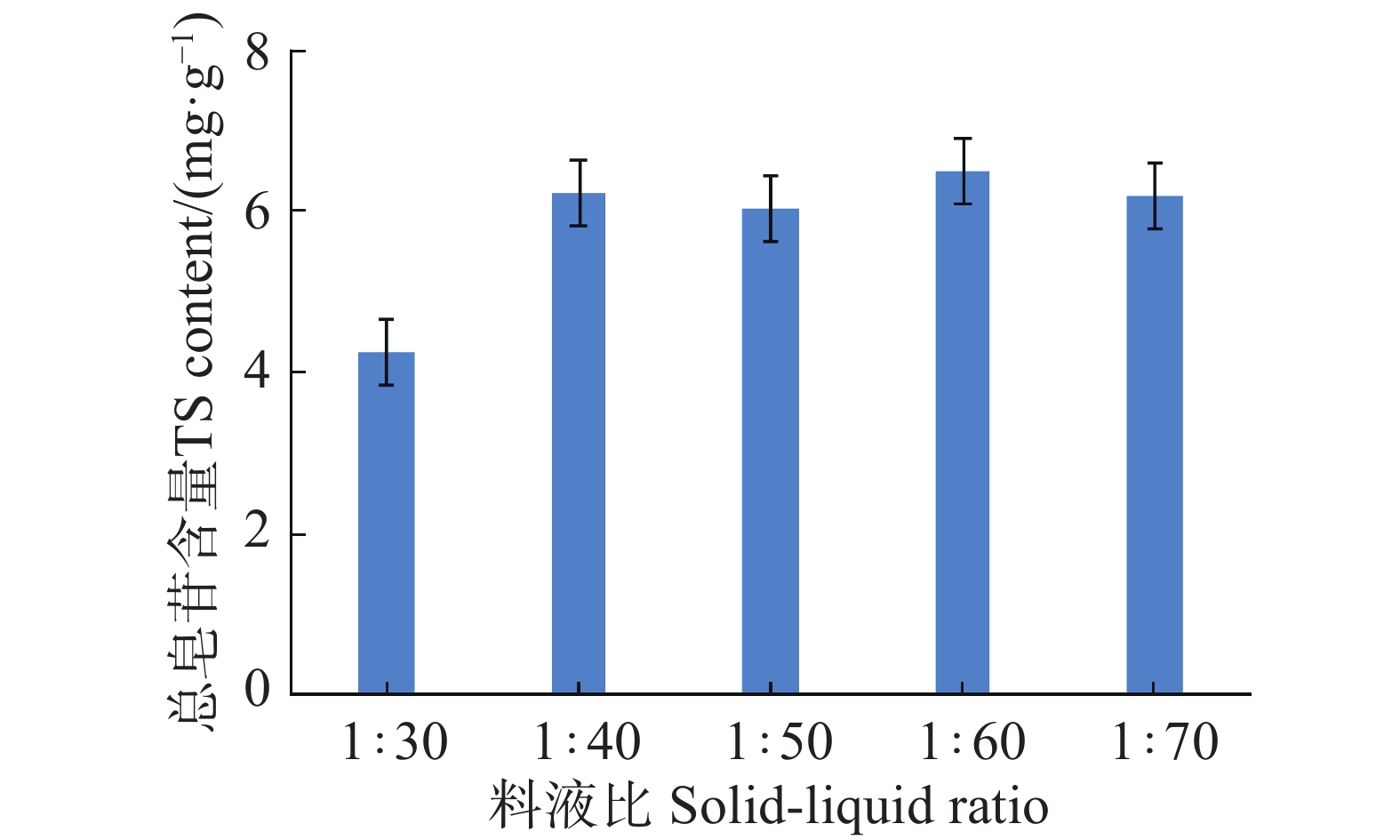
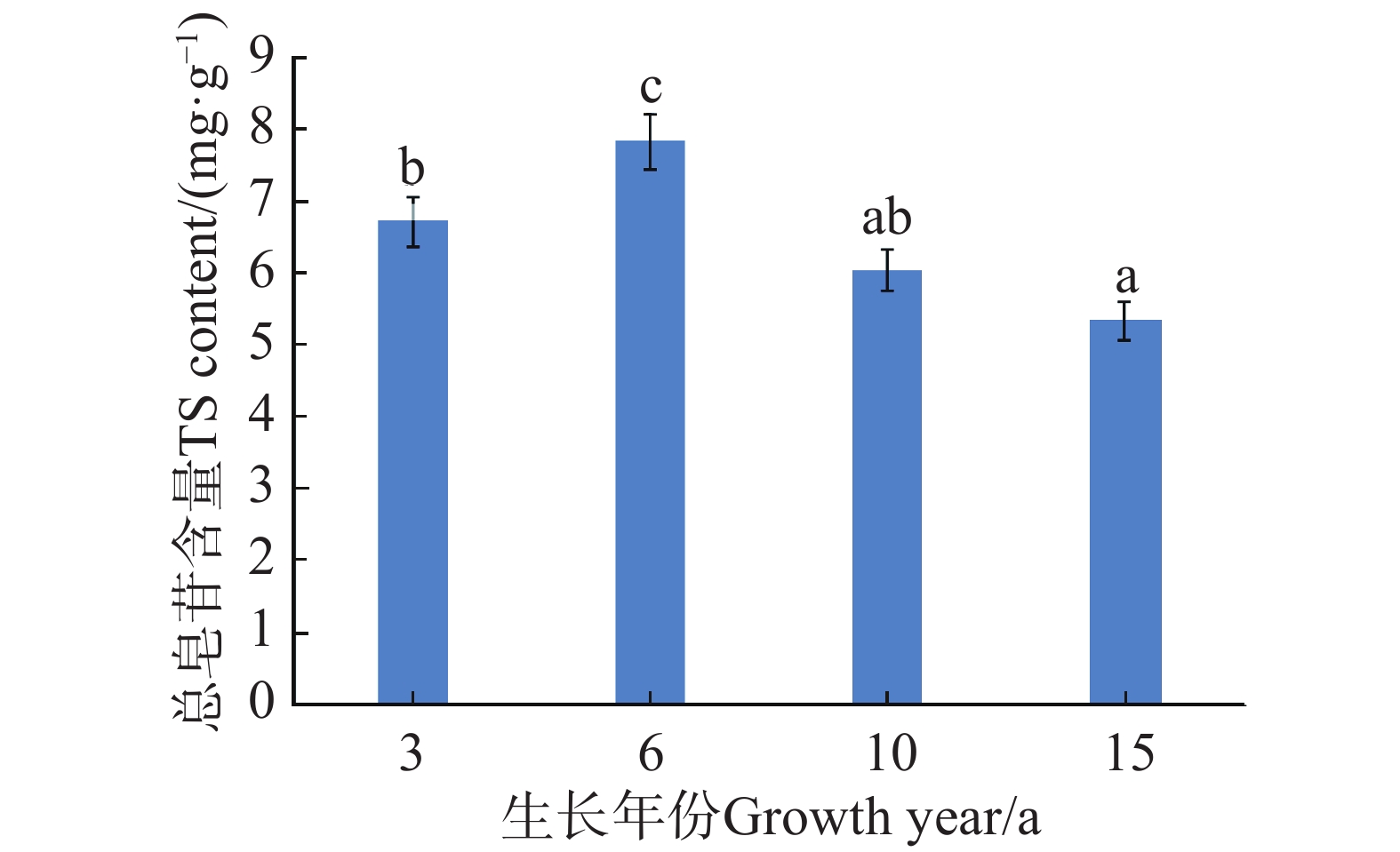
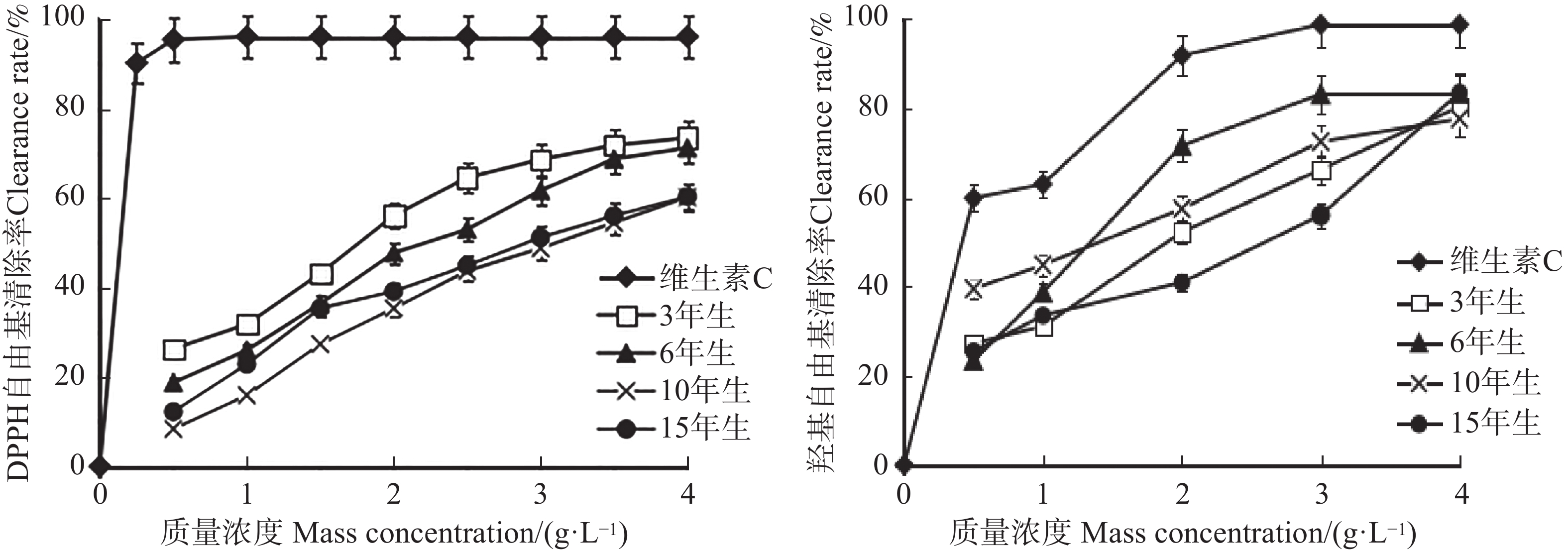
An attempt was made to determine the content of total saponins and antioxidant activity in the root of Callerya speciosa (Champion ex Bentham) Schot at different ages, and provide reference for their rational development and utilization. The root of C. speciosa were collected and extracted by using an ultrasonic-assisted extraction method, and the extraction process of total saponins from C. speciosa root were optimized. Furthermore, the total saponins content of C. speciosa root extracts from different ages (3-, 6-, 10-, and 15-year-old) was determined by using UV photometer. The antioxidant activity of total saponins from the C. speciosa root was determined by using the DPPH free radical method and hydroxyl free radical method. The results showed that the best extraction conditions for total saponins of the C. speciosa root are 80% ethanol, 1:40 solid-liquid ratio, 40 min ultrasound time, and 3 extraction times. Among the four materials, the 6-year-old C. speciosa root had the highest total saponin content, while the 15-year-old C. speciosa had the lowest. The total saponin content of the C. speciosa root tended to increase and then decrease with the ages of C. speciosa. The antioxidant activity test found that total saponins of the C. speciosa root at different ages all possessed a certain level of antioxidant capacity in vitro, which can be developed and utilized as a potential natural antioxidant.
An attempt was made to determine the content of total saponins and antioxidant activity in the root of Callerya speciosa (Champion ex Bentham) Schot at different ages, and provide reference for their rational development and utilization. The root of C. speciosa were collected and extracted by using an ultrasonic-assisted extraction method, and the extraction process of total saponins from C. speciosa root were optimized. Furthermore, the total saponins content of C. speciosa root extracts from different ages (3-, 6-, 10-, and 15-year-old) was determined by using UV photometer. The antioxidant activity of total saponins from the C. speciosa root was determined by using the DPPH free radical method and hydroxyl free radical method. The results showed that the best extraction conditions for total saponins of the C. speciosa root are 80% ethanol, 1:40 solid-liquid ratio, 40 min ultrasound time, and 3 extraction times. Among the four materials, the 6-year-old C. speciosa root had the highest total saponin content, while the 15-year-old C. speciosa had the lowest. The total saponin content of the C. speciosa root tended to increase and then decrease with the ages of C. speciosa. The antioxidant activity test found that total saponins of the C. speciosa root at different ages all possessed a certain level of antioxidant capacity in vitro, which can be developed and utilized as a potential natural antioxidant.




 Email alert
Email alert RSS
RSS