
-
Clustered Regularly Interspaced Short Palindromic Repeats-associated gene(CRISPR-Cas)全称为成簇的规律间隔的短回文重复序列,最初于1987年在大肠杆菌中发现。ISHINO Y等[1]在研究大肠杆菌iap(碱性磷酸酶)基因时,在其编码区3′端侧翼序列中发现长度为29 bp高度保守的重复核苷酸序列,重复序列的间隔为32 bp。随着对该序列的深入研究,发现该重复序列广泛存在于古细菌和细菌的基因组中,直到2002年JANSEN R正式命名该重复序列为CRISPR序列,除此之外,该研究还发现CRISPR基因的侧翼序列中有4种同源基因(CRISPR-associated gene):cas1、cas2、cas3、cas4,这些基因编码一些功能蛋白,与CRISPR有功能相关性[2]。随着深入研究,CRISPR-Cas系统的功能的免疫功能逐渐被发现,CRISPR-Cas系统类似于真核生物的RNA干扰(RNAi)[3],后经证实是细菌对噬菌体等病原生物的获得性免疫作用[4]。CRISPR-Cas系统在细菌对抗噬菌体侵入时分为3个阶段。第1阶段为适应阶段:在噬菌体侵入细菌时,Cas1-Cas2蛋白复合物根据前间隔序列临近基序(PAM)位点将噬菌体靶DNA(protospacer)切割并将这段靶DNA序列插入到CRISPR重复序列5′端的末尾,产生新的间隔序列(spacer)。第2阶段是基因的表达和处理阶段,间隔序列(spacers)和CRISPR重复序列共同进行转录,形成初转录产物pre-CRISPR RNA(pre-crRNA),后由Cas蛋白复合物对转录初产物进行切割,得到成熟的包含间隔序列(spacers)和重复序列的CRISPR RNAs(crRNAs)。不同的CRISPR-Cas系统对pre-crRNA的处理存在差异,有些由多个Cas蛋白亚基处理,有的由单个Cas蛋白处理,有的借助于宿主细胞的RNase。第3阶段为干扰阶段,在guide RNA(crRNA和tracrRNA合成的引导RNA)的引导下,利用单独Cas蛋白或是Cas蛋白复合物对靶DNA或RNA进行切割。第一类CRISPR-Cas系统在切割靶链时需要多个Cas蛋白复合体的参与,而第二类CRISPR-Cas系统在切割靶DNA或RNA时只需要单个Cas蛋白加guide RNA(gRNA)即可完成对靶链的切割。因此,第二类CRISPR-Cas系统成为现在基因编辑中重要的工具。
-
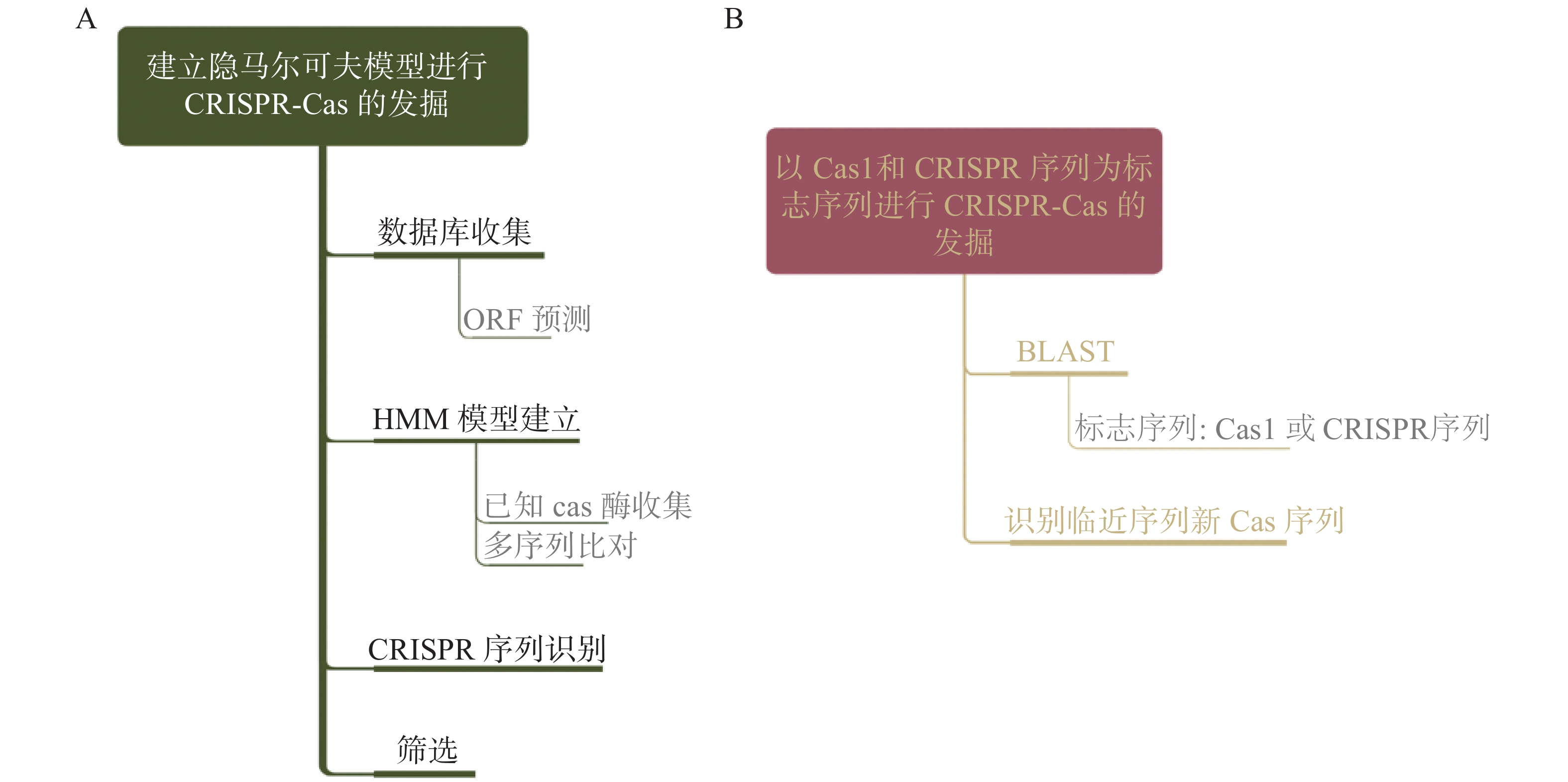
Cas蛋白作为CRISPR-Cas系统中的切割靶链的效应部分,是寻找新CRISPR-Cas系统的重点。目前,基于生物信息学手段发掘Cas系统主要分为2种方法,一种是基于对已知Cas序列建立隐马尔科夫模型(Hidden Markov Model,HMM)对细菌和古细菌基因组进行分析(图1a)。另一种是基于CRISPR-Cas系统中的标志序列对细菌和古细菌的基因组进行Cas基因的查找(图1b)。

图 1 基于生物信息学手段发掘Cas系统的2种方法
Figure 1. Two methods to explore CRISPR-Cas system based on bioinformatics
对细菌和古细菌的CRISPR-Cas系统发掘,所有的生物信息学分析都需要基于它们的基因组。CRISPR-Cas系统广泛存在于细菌和古细菌中,在收集数据时需要分为两部分。一类是基因组数据库的收集,可以通过NCBI,EBI等数据库进行细菌和古菌的全基因组数据收集和批量下载[5]。第二类是宏基因组,宏基因组由于数据库庞大,在Cas酶发掘中收集方式多样,多数通过各种野外研究发现的数据进行基因组分析[6],宏基因组数据需要组装后才可以进行下一步分析。
-
开放阅读框是指DNA序列中具有编码蛋白质潜能的序列,从起始密码子开始,终止于终止密码子。通过读取开放阅读框,可以从细菌和组装好的古细菌基因组中识别出所有可以编码蛋白的潜在基因序列,目前应用于识别原核生物基因组开放阅读框频率较高的预测软件有Prodigal[7]、Glimmer[8]和GeneMark[9]等,软件优缺点对比见表1。其中,Prodigal是在发掘新Cas酶中明确提及使用过的开放阅读框识别软件[10]。准确的识别开放阅读框并对开放阅读框的位置进行准确定位有助于后续对CRISPR序列定位后二者共同分析。
表 1 ORF预测软件对比
Table 1. ORF prediction software comparison
软件
Software优点
Advantages缺点
DisadvantagesProdigal 使用简单、所有基因组
可在同一文件运行预测结果较少 Glimmer 预测结果多 使用复杂 Genemarks 依赖自我训练集 需要单个基因组运行 -
隐马尔科夫模型是一种统计分析模型,近年来被广泛应用到各种生物信息学分析中,主要用于描述某一核苷酸序列从其特定的祖代遗传而来的概率。根据现有的序列通过计算机对序列的分析建立隐马尔科夫模型,进而推测出最有可能出现的祖代序列[11]。
在用HMMER软件建立隐马尔科夫模型之前,需要对准备建立模型的已知Cas蛋白序列进行多序列比对。目前应用于多序列比对有以下几种软件,分别为CLUSTAL W、MUSCLE、T-COFFEE、DIALING2、MAFFT等,软件速度MUSCLE最快,对比准确性MUSCLE最高[12]。通过对已知Cas酶的多序列比对,得到STOCKHOLM(sto)文件,作为接下来的模型建立输入文件。
HMMer是基于隐马尔科夫模型建立的生物信息学分析软件[13],有网页版和本地版,通过hmmbuild指令和Cas蛋白多序列比对结果输入文件建立已知Cas蛋白的隐马尔科夫模型,hmmsearch指令和建立的Cas蛋白模型输入文件可以对预测出的开放阅读框文件进行序列分析,进而推测出可能是Cas蛋白的编码序列。
-
CRISPR序列包含间隔序列和重复序列,是CRISPR-Cas系统中另一个重要的组成部分。应用于发掘CRISPR-Cas系统的目前有3种,分别为CRISPRDetect[14]、CRISPR Finder[15]和PILER-CR[16]。其中,CRISPR Finder应用最广[6, 10,17],可以准确识别出长度短的CRISPR序列,在升级后不止可以识别CRISPR序列,还可以通过自带的隐马尔科夫模型对输入的序列进行Cas蛋白的预测[18]。识别CRISPR序列软件优缺点对比见表2。
表 2 CRISPR序列识别软件对比
Table 2. Comparison of CRISPR sequence recognition software
软件
Software优点
Advantages缺点
DisadvantagesCRISPRDetect 识别序列方向 背景噪声 CRISPR Finder DRs识别及展示、
准确识别小序列单个基因
组序列运行PILER-CR 使用简单,所有基因组可放在
同一文件运行,速度快识别精度较低 -
在对基因组进行生物信息学分析后,得到软件预测出的Cas蛋白和CRISPR序列。对得到的候选序列进行筛选,筛选条件有以下3条:1)同时含有Cas1和CRISPR序列;2)与Cas1相邻的10个ORF之一包含1个大于800个氨基酸的未被鉴定的蛋白序列(通过隐马尔科夫模型预测出的);3)在同一基因组列中没有已经被鉴定出的包含Cas基因的干扰模块[10]。
-
JANSE等人的研究表明,有些CRISPR序列上下游无编码Cas的序列,有些编码Cas酶的序列上下游无CRISPR序列[2],因此,以Cas1蛋白和CRISPR序列为标志序列分别进行识别可以有效搜寻到所有候选序列。此种方法是根据已经发现的Class2 CRISPR-Cas系统的结构特征进行发掘。
-
由于Cas1序列在CRISPR-Cas系统中高度保守[19],且是在CRSIPR-Cas系统中普遍存在的编码序列,因此根据Cas1序列进行BLAST可以有效找出可能含有CRISPR-Cas系统的基因组。另一种可选的标志序列为CRISPR序列,CRISPR序列是CRISPR-Cas系统中的重要组成部分,因此也可以作为准确识别CRISPR-Cas系统的序列,为了准确识别CIRSPR序列,可以选取上述CRISPR识别软件,通过CRISPR序列找出的候选序列是通过Cas1进行序列筛选的2倍[20],这说明很多CRISPR-Cas系统是缺乏适应模块的。
-
对BLAST识别出的Cas1序列或CRISPR识别软件识别出的CRISPR序列的上下游进行分析,寻找是否有其他编码Cas蛋白。使用GeneMark软件中MetaGeneMark_v1.mod模型[20]对序列进行开放阅读框识别[21]。
对于以Cas1为标志序列识别出的序列,通过CRISPR-Cas分类标准来检查其上下游是否存在其他的Cas基因[22]。对于以CRISPR为标志识别出的序列,在识别出的CRISPR序列的上下游20 kb[23](有些研究是10 kb[23])以内识别可能编码蛋白的序列。由于Cas9蛋白和Cpf1蛋白都由大于1000个氨基酸构成[24-26],所以选择氨基酸残基大于500的编码序列(有些研究是大于700 aa[23]或750 aa[5]作为新Cas蛋白的候选序列进行下一步分析)。
确定新Cas蛋白与标志序列和CRISPR的位置关系,新Cas蛋白需要在标志序列(Cas1)的4个基因以内。多数的CRISPR-Cas系统中Cas蛋白与CRISPR序列共同出现的频率很高,限制新的Cas蛋白至少有50%[23]或70%[5]位于CRISPR序列上下游20 kb以内。
-
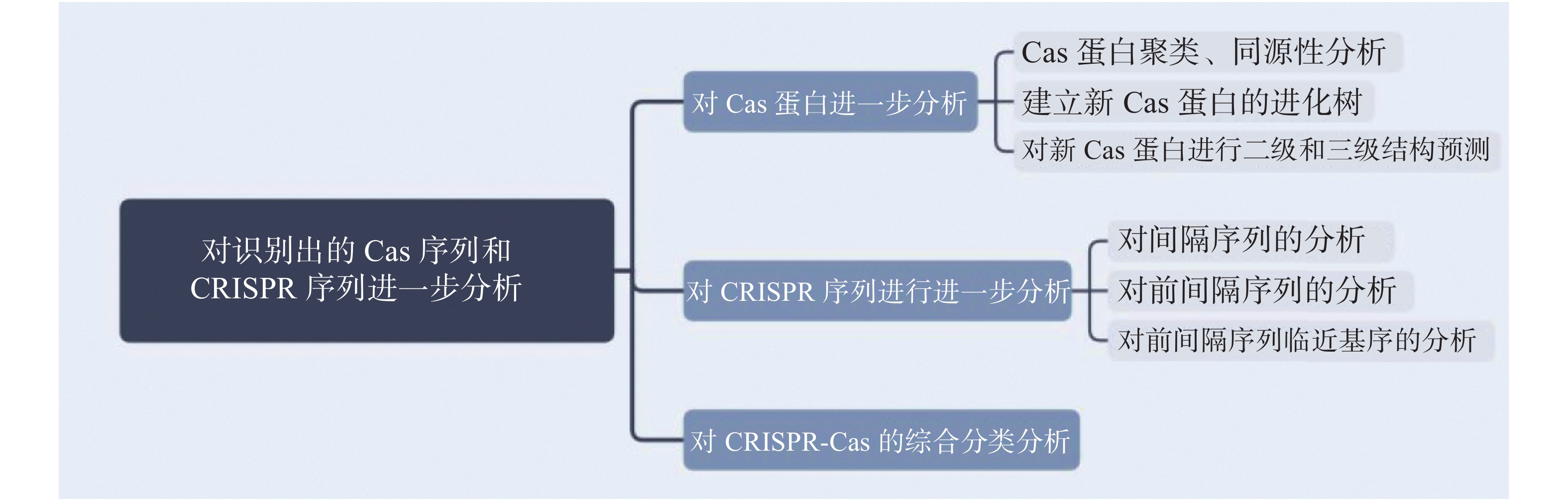
在发掘出新的CRISPR-Cas系统后,需要对其进行生物信息学分析,以便了解Cas蛋白的理化性质并进行家族分析。对CRISPR序列进行分析可以了解该系统对抗的入侵质粒噬菌体等,并有助于研究其切割位点。对识别出的Cas蛋白序列和CRISPR序列进行分析流程见图2。
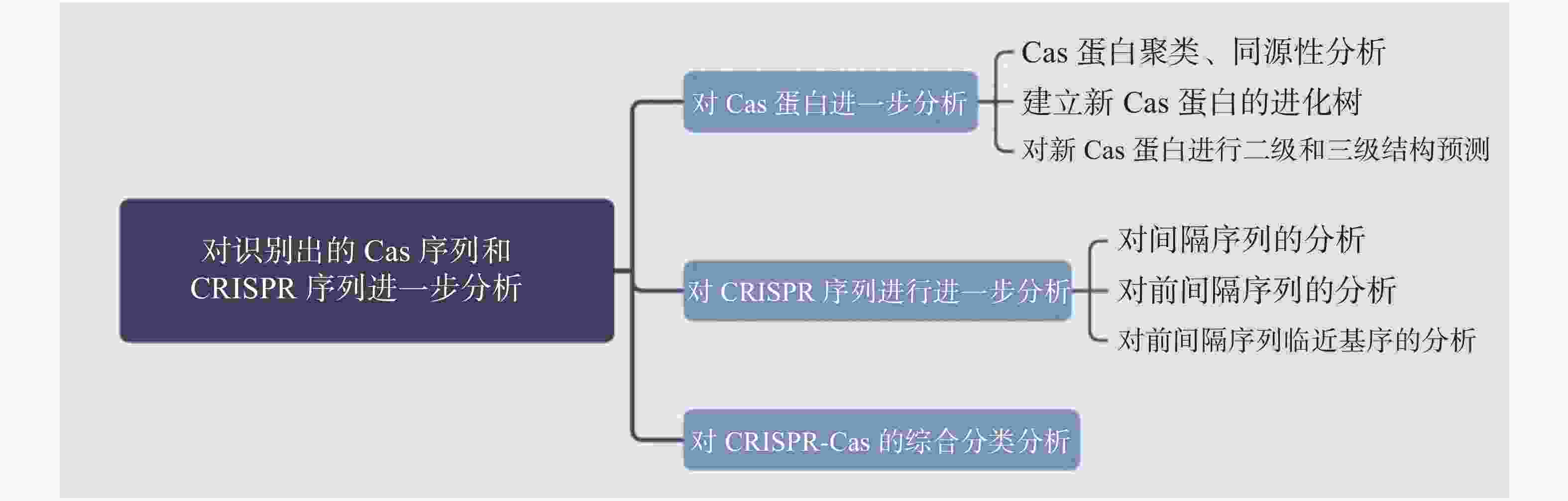
图 2 对识别出的Cas蛋白序列和CRISPR序列进行分析流程示意图
Figure 2. Schematic diagram of the analysis process of the identified Cas protein sequence and CRISPR sequence
-
对识别出的蛋白进行聚类分析,聚类分析的目的是根据已有的蛋白序列分析预测新蛋白质序列[27],并对研究蛋白质的起源和家族分析有重要意义[28],将Orthomcl[29]和MCL[30]或作为新Cas蛋白的聚类分析软件。
为了去除基因组中可能造成偏差的聚类序列,对预测出的Cas蛋白分析,通过发掘出的Cas序列和PSI-BLAST软件[31]对NCBI的非冗余(nr)蛋白和宏基因组(env_nr)蛋白数据库进行检索,利用HMM对UniProt数据库进行检索[32]可得到已知的其他同源蛋白序列[5]。使用HH-suite[33]的HHpred对发掘出的Cas蛋白进行远距离的同源蛋白检索,要求新的Cas蛋白能够检索出10个同源效应子[23]。
-
对发掘出的Cas蛋白和搜索出的同源蛋白建立进化树,比较不同Cas蛋白之间亲缘关系,是分析新发掘出的Cas蛋白的常用分析方法之一。通常进化树建立使用软件有RaxmL[34]和PhyML[35]等,上述建立进化树软件输入文件为PHYLIP(.phy)格式。再使用FigTree和iTOL[36]软件实现进化树的可视化。
-
为了进一步发掘出Cas蛋白序列特点,进行结构和结构域的预测分析。由于Cas蛋白进化速度很快,要求识别Cas蛋白结构域的软件必须能进行精确识别[19, 37]。对发掘出的Cas蛋白进行二级结构预测可以使用JPred4[38]、CD-Search[39]或HH pred[40]。蛋白质的三级结构预测软件分为同源建模法与穿线法,同源建模法预测的原理为相似的氨基酸序列对应着相似的蛋白质结构,如软件Phyre2[41]。穿线法预测通过已知蛋白的结构拓扑进行预测,不相似的蛋白也能有相似的结构,如软件I-TASSER[42]。
-
识别CRISPR序列中的间隔序列(spacers)有助于寻找对抗入侵的质粒和噬菌体。识别CRISPR序列的CRISPRFinder等软件识别出的间隔序列根据组装基因组数据确定。相关样品的短DNA或宏基因组识别间隔序列可使用CRASS软件[43]。
-
前间隔序列作为CRISPR-Cas系统进行序列切割在噬菌体或质粒上与间隔序列对应的靶序列,对前间隔序列的识别要求高相似度。查找噬菌体或质粒中的前间隔序列多使用BLAST软件中的blastn程序。针对宏基因组数据使用task blastn-short程序[5]对宏基因组组装数据库进行前间隔序列识别,要求与间隔序列(spacer)的错配碱基小于等于1,对于搜索中可能出现的CRISPR序列中的间隔序列干扰,通过其重复性去除。除此之外,还可使用megablastn[44]程序,对病毒的非冗余数据库和原核生物基因组数据库进行搜索。此方法要求前间隔序列与间隔序列长度L最大错配数限制在区间(0,√l−22)[20]。
-
前间隔序列临近基序(PAMs),是一些Class2 CRISPR-Cas系统,如Cas9蛋白识别靶序列的识别位点,通常在靶DNA的3′末端作用,有研究猜测PAMs与DNA解旋作用有关[45]。PAMs的识别通过前间隔序列侧翼序列的对齐区域进行查找,PAMs的可视化和DNA图形展示通过WebLogo[46]软件进行。在前间隔序列和间隔序列的对齐过程中,如果出现一个间隔序列与多个不同位置的具有不同侧翼序列前间隔序列匹配,则前间隔序列和下游核苷酸的每一种不同组合都应考虑进PAMs的计算中[5]。
-
为了准确分析发掘出的CRISPR-Cas系统和新的Cas蛋白,在对其进行进一步分析前,应根据新的CRISPR-Cas系统进行分类,CRISPR-Cas系统分类可根据近期发表的分类方法进行[22],根据不同type和subtype的标志基因,如Cas3、Cas9和Cas12等对识别出的>500 aa的CRISPR-Cas系统进行分类。
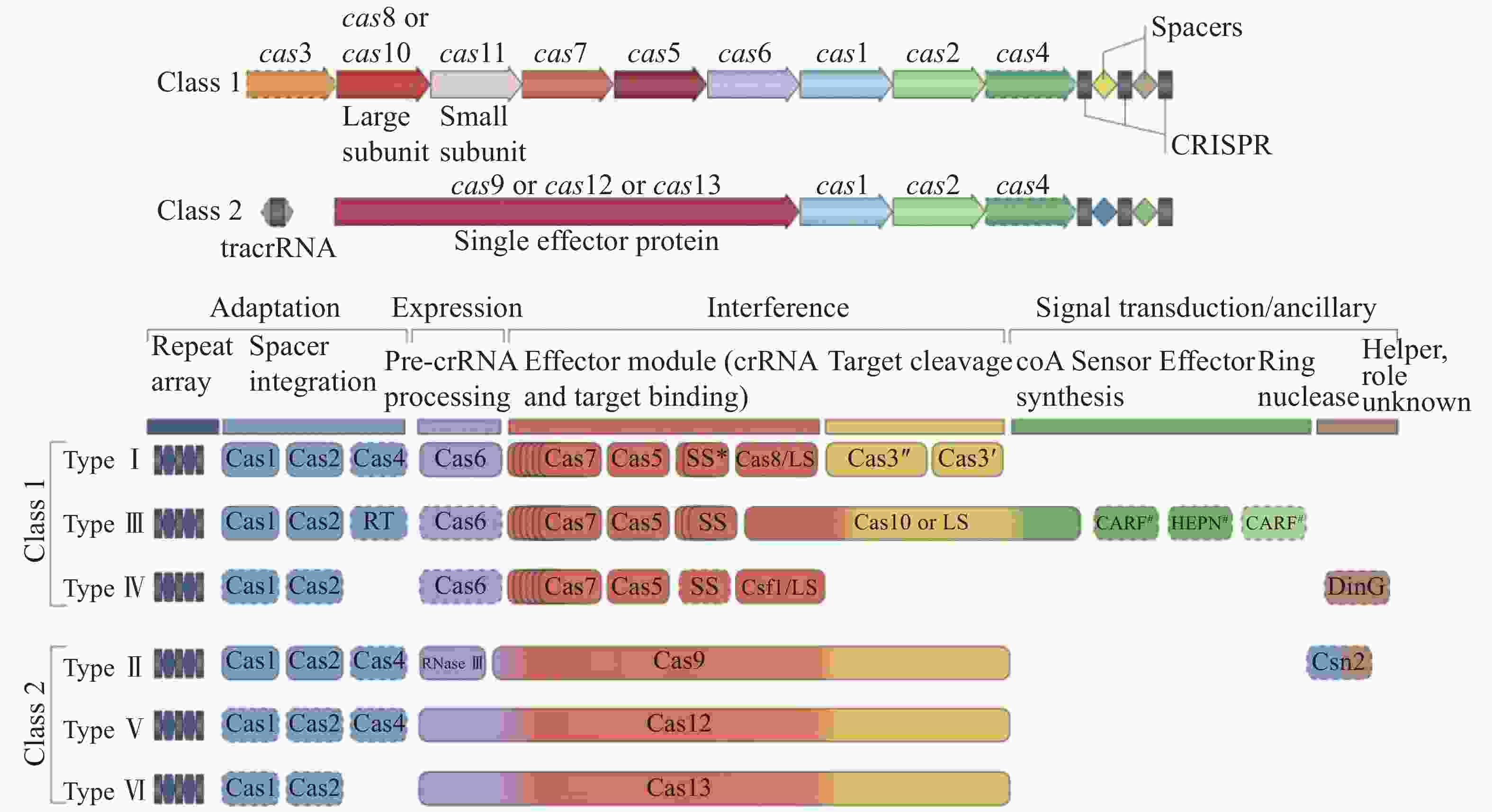
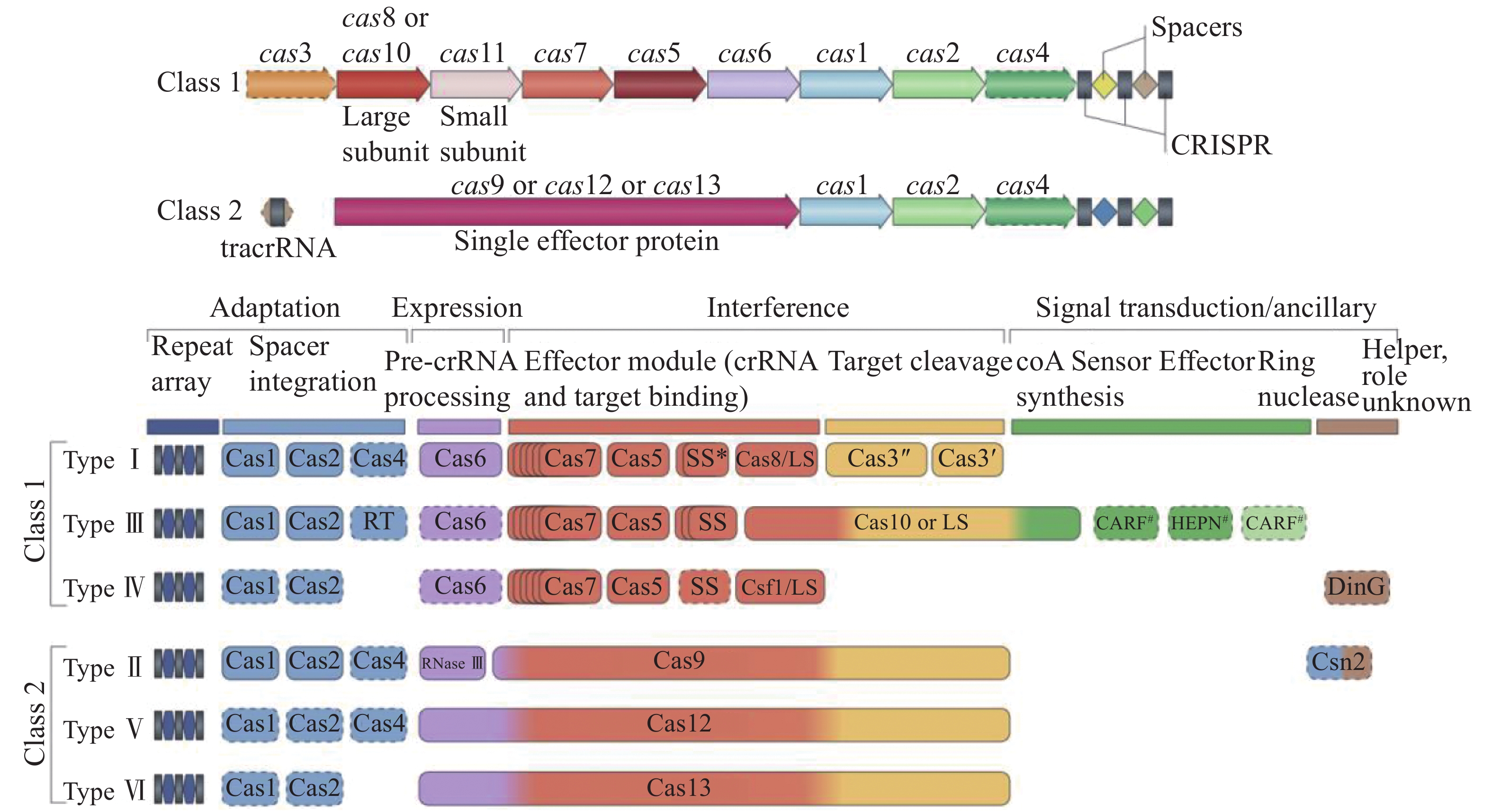
CRISPR-Cas系统分类方法有根据获得模块(Cas1-Cas2)进行分类、根据CRISPR的序列相似性或结构相似性进行分类、根据Cas1发生进行分类、根据CRISPR-Cas基因座结构分类、根据效应模块进行分类、根据亚型分类、根据物种分类。MAKAROVA等2015年的研究对比了不同CRISPR-Cas系统分类方法的不同(图3),结果表明,通过效应模块进行CRISPR-Cas系统分类通过蛋白质的相似性能在聚类处理后的库中搜寻到更多结果,通常能够直接对应已经发现的各种亚型[22]。因此,MAKAROVA等人基于效应模块建立了一种CIRSPR-Cas系统的自动注释的方法。Cas1-Cas2组成的获得模块作为最普遍的序列未被选择的原因是其虽与Cas1系统发育树密切相关,但与CRISPR-Cas基因座结构相关性弱。他们建立的这种分类方法的精确度能达到0.998。

CRISPR-Cas系统分为两大类(图4):一类(Class1)是多个Cas蛋白与crRNA共同作用切割把链的CRISPR-Cas系统,另一类(Class2)是以Cas9为代表的单亚基与crRNA共同作用切割靶链的作用系统。目前的分类方法根据不同的特征基因将Cas蛋白分为6种类型,其中Class1分为3种类型,Type Ⅰ:以Cas3或Cas3基因的变异体为标志基因,在细菌和古细菌中都有广泛分布;Type Ⅲ:以Cas10基因为标志基因,编码多亚基蛋白并包含一个RNA识别区域,Type Ⅲ在细菌和古菌中也都有分布;Type Ⅳ,缺少编码Cas1-Cas2基因,且部分编码蛋白远离CRISPR序列,此种蛋白多分布于细菌中。Class2分为3种类型:Type Ⅱ:以Cas9基因为标志基因,在细菌和古菌中都有分布;Type Ⅴ:以Cas12和Cas14基因为标志基因,临近Cas1-Cas2和CRISPR序列,并与TnpB蛋白有高度相似性,此种类型大多数分布于细菌中;Type Ⅵ:以Cas13位标志基因[22, 47]。
新的分类和命名方法根据序列相似性、同源序列分析和上下游序列比较进行CRISPR-Cas系统的分类。Class2中,包含了种类Ⅱ、种类Ⅴ和种类Ⅵ及他们的变异体(最新分类),其中type Ⅱ的Cas9蛋白包含HNH和RuvC-like两种结构域,分别切割靶DNA的两条链。typeV的Cas12蛋白只包含RuvC-like结构域负责切割DNA的两条链。TypeⅥ的Cas13蛋白包含2个HEPN结构域,除此之外,还有非特异性的核糖核酸酶活性。
-
笔者以生物信息学手段为重点,将基于微生物基因组CRISPR-Cas系统发掘细分为:1)基于隐马尔科夫模型的发掘方法:i)开放阅读框预测,ii)收集已知的Cas蛋白建立隐马尔科夫模型,iii)CRISPR序列识别;2)以Cas1和CRISPR为标志序列进行CRRISPR-Cas发掘:i)通过标志序列Cas1或CRISPR序列对基因组进行检索,ii)对标志序列的上下游蛋白进行分析寻找可能存在的Cas酶。提供了在识别出新CRISPR-Cas系统后,对新CRISPR-Cas系统的Cas酶进行的聚类分析(BLAST、HHpred等软件)、进化树建立(RAxml等软件)、结构域和三级结构预测(JPred4等软件)分析方法;3)对新CRISPR-Cas系统中,CRISPR序列的间隔序列(CRASS等软件)、前间隔序列(blastn等)前间隔序列临近基序分析。
然而,不同的分析方法在实践应用中会有相应的限制。Cas酶发掘方面,通过隐马尔科夫建立模型的手段只能根据已知的Cas酶预测出与已知相似的类型,不能预测出序列差别大的两种不同类型Cas蛋白。通过标志序列Cas1和CRISPR序列进行的新Cas酶发掘对CRISPR-Cas系统的结构有严格要求,发掘出的CRISPR-Cas系统必须在上下游20 kb以内含有标志序列。随着新发现的Class2 CRISPR-Cas14中Cas蛋白只有400~700个氨基酸[6],传统认为,单个蛋白可以产生靶向切割作用的Cas蛋白需要大于950个氨基酸残基的观点被颠覆,因此,对于标志基因上下游>700氨基酸残基的蛋白筛选限制条件应当更新。此外,Cas蛋白进化分类方面随着Cas12发现可能与TnpB蛋白转座有关,提供了不同Cas蛋白起源不同的新观点。CRISPR序列识别方面,有些软件并不能展示出DR序列或是序列方向,因此,可能会造成PAM分析和结构分析的误差。
CRISPR系统分类上看,随着近年来CRISPR-Cas系统研究的不断发展,分类方法应不断更新。主要原因如下:1)随着CRISPR-Cas生物信息学发掘工具的不断发展,已经发现靶RNA切割的Ⅵ型和Ⅴ型CRISPR-Cas系统,并有个Ⅴ型的亚型被发现。有研究表明,Ⅴ型CRISPR-Cas系统是从转座子TnpB核酸酶通过基因座转移和重复进化产生,因此Ⅴ型CRISPR-Cas系统出现了大量的突变体,并且有相当一部分进化成了独立的亚型[48]。2)近年来发现的CRISPR-Cas系统中,被认为在细菌或古菌中执行不同于获得性免疫的功能[49],不含有靶链切割的能力,这些被认为功能不同的CRISPR-Cas序列通常编码于转座子等可以动的编码区中[48,50]。3)多种涉及到CRISPR-Cas系统的标志基因与信号传递和调控作用有关[51-52]。
CRISPR-Cas系统作为定向基因编辑的革命性技术,拥有巨大的潜力和广阔的研究前景。已经发现的Class2 CRISPR-Cas系统可以定向切割靶单链DNA/RNA和靶双链DNA,然而,至今为止尚未有科学家发现可切割双链RNA的CRISPR-Cas系统。随着越来越多的微生物和宏基因组数据被提供、越来越精进的基因组测序以及不断完善的生物信息学方分析法,会有更多的CRISPR-Cas系统被发现并应用于基因组的定向编辑,帮助人们了解分析动植物基因功能。
Methods for Discovery and Analysis of Class2 CRISPR-Cas Systems
-
摘要: 近年来,规律间隔成簇短回文系统(CRISPR-Cas)作为基因编辑手段在动植物基因编辑中已广泛应用。现已被证实的Class2类CRISPR-Cas系统CRISPR-Cas12、CRISPR-Cas14等均通过生物信息学手段被发掘出来,因此,生物信息学成为发现新CRISPR-Cas系统及其子类型的重要方法。笔者综述了Cas酶两类生物信息学发掘手段,一类方法是通过已知Cas酶建立隐马尔科夫模型(HMM)预测可能的同类Cas酶;另一类方法是以标志序列Cas1或CRISPR识别为基础分析上下游可能的Cas酶,同时讨论了两种方法的限制。在此基础上,综述了Cas蛋白和CRISPR序列进一步分析方法,包括Cas蛋白同源性、进化分析及CRISPR序列间隔序列(spacers)、前间隔序列(protospacers)前间隔序列临近基序(PAM)分析。
-
关键词:
- Cas酶发掘 /
- CRISPR-Cas系统 /
- 生物信息学分析
Abstract: Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR-Cas) has been widely used as a tool in recent years for gene editing in animal and plant gene editing. The proven Class2 CRISPR-Cas systems, such as CRISPR-Cas12 and CRISPR-Cas14, have been discovered through bioinformatics mining. Bioinformatics has become an important tool for discovering of new CRISPR-Cas systems and their subtypes. Two methods for bioinformatics mining of Cas enzymes are reviewed. One method is to create a hidden Markov model (HMM) using known Cas enzymes to predict similar Cas enzymes, and the other method is to analyze the possible upstream and downstream Cas enzymesbased on the recognition of the marker sequence Cas1 or CRISPR. The limitations of these two methods are discussed. Furthermore, methods for further analysis of Cas protein and CRISPR sequences are also reviewed, including Cas protein homology, phylogenetic analysis, and analysis of CRISPR sequence spacers, protospacers&protospacer adjacent motifs (PAM).-
Key words:
- mining of Cas enzyme /
- CRISPR-Cas system /
- bioinformatics analysis
-
图 2 对识别出的Cas蛋白序列和CRISPR序列进行分析流程示意图
Fig. 2 Schematic diagram of the analysis process of the identified Cas protein sequence and CRISPR sequence
表 1 ORF预测软件对比
Table 1 ORF prediction software comparison
软件
Software优点
Advantages缺点
DisadvantagesProdigal 使用简单、所有基因组
可在同一文件运行预测结果较少 Glimmer 预测结果多 使用复杂 Genemarks 依赖自我训练集 需要单个基因组运行  下载: 导出CSV
下载: 导出CSV
表 2 CRISPR序列识别软件对比
Table 2 Comparison of CRISPR sequence recognition software
软件
Software优点
Advantages缺点
DisadvantagesCRISPRDetect 识别序列方向 背景噪声 CRISPR Finder DRs识别及展示、
准确识别小序列单个基因
组序列运行PILER-CR 使用简单,所有基因组可放在
同一文件运行,速度快识别精度较低
下载: 导出CSV
-
[1] ISHINO Y, SHINAGAWA H, MAKINO K, et al. Nucleotide sequence of the iap gene responsible for alkaline phosphatase isozyme conversion in Escherichia coli and identification of the gene product [J]. Journal of Bacteriology, 1987, 169(12): 5429 − 5433. doi: 10.1128/JB.169.12.5429-5433.1987 [2] JANSEN R, EMBDEN J D, GAASTRA W, et al. Identification of genes that are associated with DNA repeats in prokaryotes [J]. Molecular Microbiology, 2002, 43(6): 1565 − 1575. doi: 10.1046/j.1365-2958.2002.02839.x [3] MAKAROVA K S, GRISHIN N V, SHABALINA S A, et al. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action [J]. Biology Direct, 2006, 1(7): 1 − 26. [4] BARRANGOU R, FREMAUX C, DEVEAU H, et al. CRISPR provides acquired resistance against viruses in prokaryotes [J]. Science, 2007, 315(5819): 1709 − 1712. doi: 10.1126/science.1138140 [5] KONERMANN S, LOTFY P, BRIDEAU N J, et al. Transcriptome engineering with RNA-targeting type Ⅵ-D CRISPR e ffectors [J]. Cell, 2018, 173(3): 665 − 676. doi: 10.1016/j.cell.2018.02.033 [6] LUCAS B H, DAVID B, JANICE S C, et al. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes [J]. Science, 2018, 362(6416): 839 − 842. doi: 10.1126/science.aav4294 [7] HYATT D, CHEN G L, LOCASCIO P F, et al. Prodigal: prokaryotic gene recognition andtranslation initiation site identification [J]. Bmc Bioinformatics, 2010, 11(119): 1 − 11. [8] DELCHER A L, BRATKE K A, POWERS E C, et al. Identifying bacterial genes and endosymbiont DNA with Glimmer [J]. Bioinformatics, 2007, 23(6): 673 − 679. doi: 10.1093/bioinformatics/btm009 [9] BESEMER J, LOMSADZE A, BORODOVSKY M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions [J]. Nucleic Acids Research, 2001, 29(12): 2607 − 2618. doi: 10.1093/nar/29.12.2607 [10] BURSTEIN D, HARRINGTON L B, STRUTT S C, et al. New CRISPR-Cas systems from uncultivated microbes [J]. Nature, 2017, 542(7640): 237 − 241. doi: 10.1038/nature21059 [11] 周海廷. 隐马尔科夫过程在生物信息学中的应用[J]. 生命科学研究, 2002, 6(3): 204 − 210. doi: 10.3969/j.issn.1007-7847.2002.03.004 [12] WONG K M, SUCHARD M A, HUELSENBECK J P. Alignment Uncertainty and Genomic Analysis [J]. Science, 2008, 319(5862): 473 − 476. doi: 10.1126/science.1151532 [13] POTTER S C, LUCIANI A, EDDY S R, et al. HMMER web server: 2018 update [J]. Nucleic Acids Research, 2018(46): 200 − 204. [14] BISWAS A, STAALS J, MORALES S E, et al. CRISPRDetect: A flexible algorithm to define CRISPR arrays [J]. BMC Genomics, 2016, 17(1): 1 − 14. [15] IBTISSEM G, GILLES V, CHRISTINE P. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats [J]. Nucleic Acids Research, 2007(35): 52 − 57. [16] Robert C E. PILER-CR: Fast and accurate identification of CRISPR repeats [J]. BMC Bioinformatics, 2007, 8(18): 1 − 6. [17] ZETSCHE B, GOOTENBERG J S, ABUDAYYEH O O, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cassystem [J]. Cell, 2015(163): 1 − 13. [18] COUVIN D, BERNHEIM A, TOFFANO-NIOCHE C, et al. CRISPRCasFinder, an update of CRISRFinder, includes a portable version enhanced performance and integrates search for Casproteins [J]. Nuclc Acids Research, 2018(46): 246 − 251. [19] TAKEUCHI N, WOLF Y I, MAKAROVA S, et al. Nature and intensity of selection pressure on CRISPR-associated genes [J]. Journal of Bacteriology, 2011, 194(5): 1216 − 1225. [20] SHMAKOV S, SMARGON A, SCOTT D, et al. Diversity and evolution of class 2 CRISPR–Cassystems [J]. Nature Reviews Microbiology, 2017, 15(3): 169 − 182. doi: 10.1038/nrmicro.2016.184 [21] WENHAN ZHU, LOMSADZE A, BORODOVSKY M. Ab initio gene identification in metagenomic sequences [J]. Nucleic Acids Research, 2010, 38(12): e132. doi: 10.1093/nar/gkq275 [22] MAKAROVA K S, WOLF Y I, ALKHNBASHI O S, et al. An updated evolutionary classification of CRISPR-Cassystems [J]. Nature Reviews Microbiology, 2015, 13(3569): 722 − 736. [23] SMARGON A A, COX D B, PYZOCHA N K, et al. Cas13b is a type VI-B CRISPR-associated RNA-guided RNasedifferentially regulated by accessory proteins Csx27 and Csx28 [J]. Molecular Cell, 2017(65): 618 − 630. [24] NISHIMASU H, RAN A F, PATRICK D H, et al. Crystal structure of Cas9 in complex with guide RNA and target DNA [J]. Cell, 2014(156): 935 − 949. [25] NISHIMASU H, CONG L, YAN W, et al. Crystal structure of Staphylococcus aureusCas9 [J]. Cell, 2015, 162(5): 1113 − 1126. doi: 10.1016/j.cell.2015.08.007 [26] YAMANO T, NISHIMASU H, ZETSCHE B, et al. Crystal structure of Cpf1 in complex with guide RNA and target DNA [J]. Cell, 2016, 165(4): 949 − 962. doi: 10.1016/j.cell.2016.04.003 [27] 唐东明, 朱清新, 陈科, 等. 一种有效的蛋白质序列聚类分析方法[J]. 软件学报, 2011, 22(8): 1827 − 1837. [28] YING ZHAO, KARYPIS G. Data clustering in life sciences [J]. Molecular Biotechnology, 2005, 31(1): 55 − 80. doi: 10.1385/MB:31:1:055 [29] LI L. OrthoMCL: Identification of orthologgroups for eukaryotic genomes [J]. Genome Research, 2003, 13(9): 2178 − 2189. doi: 10.1101/gr.1224503 [30] ENRIGHT A J, DONGEN S V, OUZOUNIS C A. An efficient algorithm for large-scale detection of protein families [J]. Nucleic Acids Research, 2002, 30(7): 1575 − 1584. doi: 10.1093/nar/30.7.1575 [31] ARON M B, PANCHENKO A R, SHOEMAKER B A, et al. CDD: a database of conserved domain alignments with links to domain three-dimensional structure [J]. Nucleic Acids Research, 2002(30): 281 − 283. [32] UNIPROT C. The UniProt Consortium. UniProt: a hub for protein information [J]. Nucleic Acids Research, 2015, 43(D1): D204 − D212. doi: 10.1093/nar/gku989 [33] REMMERT M, BIEGERT A, HAUSERA, et al. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment [J]. Nature Methods, 2011, 9(2): 173 − 175. [34] ALEXANDROS S. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies [J]. Bioinformatics, 2014(9): 1312 − 1313. [35] GASCUEL O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0 [J]. Systematic Biology, 2010, 59(3): 307 − 321. doi: 10.1093/sysbio/syq010 [36] IVICA L, PEER B. Interactive Tree of Life (iTOL): An online tool for phylogenetic tree display and annotation[M]. New York: Oxford University Press, 2007. [37] MAKAROVA K S, WOLF Y I, KOONIN E V. Comparative genomics of defense systems in archaea and bacteria [J]. Nucleic Acids Research, 2013, 41(8): 4360 − 4377. doi: 10.1093/nar/gkt157 [38] Alexey D, Christian C, James P, et al. JPred4: a protein secondary structure prediction server [J]. Nucleic Acids Research, 2015, 43(332): 389 − 394. [39] MARCHLER-BAUER A, STEPHEN H B. CDD: conserved domains and protein three-dimensional structure [J]. Nucleic Acids Research, 2004, 32(454): 327 − 331. [40] SODINGJ. Protein homology detection by HMM-HMM comparison. [J]. Bioinformatics, 2005(21): 951 − 960. [41] KELLEY L A, MEZULIS S, YATES C M, et al. The Phyre2 web portal for protein modeling, prediction and analysis [J]. Nature Protocol, 2015, 10(6): 845 − 858. doi: 10.1038/nprot.2015.053 [42] ROY A, KUCUKURAL A, ZHANG Y. I-TASSER: a unified platform for automated protein structure and function prediction [J]. Nature Protocols, 2010, 5(4): 725 − 738. doi: 10.1038/nprot.2010.5 [43] SKENNERTON C T, MICHAEL I, TYSON G W. Crass: identification and reconstruction of CRISPR from unassembled metagenomicdata [J]. Nucleic Acids Research, 2013, 41(10): 105. doi: 10.1093/nar/gkt183 [44] ZHANG Z, SCHWARTZ S, WAGNER L, et al. A greedy algorithm for aligning DNA sequences. [J]. Journal of Computational Biology, 2000, 7(2): 203 − 214. [45] JINEK M, CHYLINSKI K, FONFARA I, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity [J]. Science, 2012, 337(6096): 816 − 821. doi: 10.1126/science.1225829 [46] GAVIN E C, GARY H, JOHN J M, et al. WebLogo: a sequence logo generator [J]. Genome Research, 2004, 14(6): 1188 − 1190. doi: 10.1101/gr.849004 [47] MAKAROVA K S, WOLF Y I, IRANZO J, et al. Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants [J]. Nature Reviews Microbiology, 2020, 18(2): 67 − 83. doi: 10.1038/s41579-019-0299-x [48] KOONIN E V, MAKAROVA K S. Mobile genetic elements and evolution of CRISPR-Cassystems: all the way there and back [J]. Genome Biology and Evolution, 2017, 9(10): 2812 − 2825. doi: 10.1093/gbe/evx192 [49] GUILHEM F K, MAKAROVA K S, KOONIN E V. CRISPR-Cas: complex functional networks and multiple roles beyond adaptive immunity [J]. Journal of Molecular Biology, 2019, 4(431): 3 − 20. [50] PETERS J E, MAKAROVA K S, SHMAKOV S, et al. Recruitment of CRISPR-Cas systems by Tn7-like transposons [J]. Proceedings of the National Academy of Sciences, 2017, 114(35): 7358 − 7366. doi: 10.1073/pnas.1709035114 [51] MIGLE K, GEORGIJ K, CESLOVAS V, et al. A cyclic oligonucleotide signaling pathway in type III CRISPR-Cassystems [J]. Science, 2017(357): 605 − 609. [52] NIEWOEHNER O, GARCIA-DOVAL C, ROSTOL J T, et al. Type III CRISPR-Cas systems produce cyclic oligoadenylate second messengers [J]. Nature, 2017, 548(7669): 543 − 548. doi: 10.1038/nature23467 -
 点击查看大图
点击查看大图
图(4) / 表 (2)
计量
- 文章访问数: 816
- HTML全文浏览量: 704
- PDF下载量: 97
- 被引次数: 0