

-
人类活动导致的生境破碎化是全球生物多样性面临的主要威胁之一[1]。破碎化对植物种群的影响表现为减少花粉和种子的传播,增加基因漂变和近亲繁殖的风险,并导致遗传多样性降低[2−3]。对于小种群而言,破碎化使潜在配偶数量减少,从而可能增加双亲间近亲繁殖甚至自交的发生概率。长期而言,破碎化可能通过积累遗传负荷引发近交抑制,若种群无法抵抗这种负面效应,将进一步增加灭绝风险[4−5]。
兰科植物是最多样化的植物类群之一,但由于生物学特化、对栖息地的特殊偏好,在生境破碎化的影响下,兰科植物也是消失最快的植物类群之一。这种高特化性主要体现在其与传粉者及真菌共生体之间的紧密依赖关系。兰科植物的繁殖往往依赖特定的传粉者,任何传粉网络的破裂或关键物种的缺失,都会导致结实率显著下降[6]。此外,其极细小、缺乏胚乳的种子在萌发阶段高度依赖特定类型的菌根真菌[7],使得定殖和更新过程对微生境条件极为敏感。因此,在破碎化景观中,兰科植物不仅面临生境面积减少的直接压力,还容易因传粉者流失和菌根真菌分布受限而导致繁殖受阻与基因流减少[8]。这些生态过程的连锁反应可能最终造成种群规模缩小、遗传多样性下降以及局部灭绝风险的增加[9]。在此背景下,从遗传多样性的角度探讨生境破碎化对兰科植物的影响,对于理解其脆弱性的生态与遗传机制,并为保护与恢复提供理论依据,具有重要意义[10]。
海南岛的土地利用格局自20世纪80年代以来发生了显著变化,橡胶Hevea brasiliensis、槟榔Areca catechu等经济林种植以及道路和建设用地持续扩张,导致低海拔热带森林和风水林不断被侵蚀和切割[11]。在海南热带雨林国家公园中分布有一些黎族苗族传统村落,由于传统习惯和图腾崇拜,这些传统村落往往保留了以桑科植物(如见血封喉Antiaris toxicaria、高山榕Ficus altissima、黄葛树Ficus virens等)、木棉Bombax ceiba、椰子Cocos nucifera、杧果Mangifera indica等为主的“风水林”,体现了人与自然完美适应的传统生活方式,是热带地区岛屿性部族聚落的典型代表。这些传统遗留乔木为海南钻喙兰(Rhynchostylis gigantea)、短序脆兰(Acampe papillosa)和钗子股(Luisia morsei)等附生兰提供了相对安全稳定的“避难所”,使得从原始森林溢出的种群能够保存并延续下来,形成了人居环境下的种群分布。然而由于村庄之间固有的距离间隔以及不断地建设开发,导致附生兰种群出现一定程度的破碎化现象,这种破碎化是否会对附生兰种群的遗传多样性产生显著影响,以及传统“风水林”作为特殊生境在维持其遗传连接性和种群稳定性中扮演何种角色,目前尚缺乏系统性的研究。
海南钻喙兰是一种典型的热带附生兰科植物,通常可存活10年以上,寿命长、更新速率慢,使其对栖息地破碎化更加敏感,其种群主要分布于海南中部、西南部山区以及村落周边[12−13]。在人居环境的“风水林”中形成的海南钻喙兰种群,为研究破碎化生境下珍稀附生兰的遗传适应机制提供了独特的天然实验体系。本研究以传统村落中的海南钻喙兰种群为研究对象,采用SNP分子标记技术[14−15],通过分析种群的遗传多样性水平、遗传结构特征及系统发育关系,揭示破碎化对其遗传多样性的影响,为制定科学合理的保护策略提供理论依据。
-
本研究地点位于海南岛西南部昌江黎族自治县的七叉镇(19°06′ N,109°03′ E),地势结构复杂,以山地类型为主。样地海拔高度为20~200 m。该地区气候类型为典型的热带季风气候,全年四季变化不明显。年平均气温在21℃至24℃之间,年平均降雨量约为
1574 mm,干湿季分明,雨季主要集中在每年7月至10月,全年平均相对湿度约为84.2%。土壤类型主要以山地砖红壤和黄壤为主。3个海南钻喙兰种群(B、M、J)均位于村庄周边,附生于传统遗留的乔木木棉(Bombax ceiba)或杧果(Mangifera indica)上,3个种群之间的地理距离为678.5~1 469.0 m。现场调查显示,B~M 2个种群之间仍保留部分连续乔木,可作为残存扩散通道;而M~J之间的区域被道路、农田及槟榔园隔离,仅存零散宿主树,生态连通性显著降低。由于不同风水林斑块中海南钻喙兰的实际数量存在差异,导致3个采样群体的样本量不一致。B群体所在的面积较大,宿主乔木数量丰富,实际植株数量最多,因此采集了46个样本;M和J群体所在斑块面积较小,宿主资源有限,且部分植株因人为采集而使数量减少,因此,仅采集到11和15个样本。所有样本均按照“尽可能覆盖全部可见植株”的原则进行采集。
-
采集了来自B、M、J 3个种群共72个植株的新鲜健康叶片,放入硅胶中快速干燥并转移至-80℃冰箱中低温储存。使用改良的CTAB方法提取总DNA[16]。样品DNA完整性检测采用1.2%(m/m)的琼脂糖凝胶电泳,浓度检测采用qubit3.0荧光定量仪,对于降解严重样品采用主带marker亮度定量。对每1个合格的DNA样品进行等体积等浓度调整,调整完后的体积为10 μL,DNA总量为200 ng。在每个调整好浓度体积的样品DNA中,加入10 μL预混的双酶切(NEB产EcoR I + Msp I,加入量依据酶活而定)混合液,用20 μL枪头充分吸打混匀。然后放入PCR仪上进行酶切处理,设定温度为37℃反应8 h、65℃反应20 min、12℃保温。吸取每样酶切产物5 μL进行琼脂糖凝胶电泳检测,无明显total DNA主带且酶切产物在泳道中呈现一条完整的弥散条带即可判断酶切完全。在每个样品的酶切产物中加入带有特定标签(barcode)的EcoR I端接头后,再加入含有通用Msp I接头的连接混合液(连接采用NEB产T4 DNA 连接酶),用200 μL枪头充分吸打混匀。然后放入PCR仪上进行酶切处理,设定温度为16℃反应8 h、65℃反应20 min、12℃保温。连接完成后,将带有不同barcode的样品酶切/连接产物等体积混合,然后进行琼脂糖凝胶电泳下的切胶回收(Omega胶回收试剂盒),筛选片段范围为400~600 bp。对回收后的文库进行文库扩增以达到上机测序浓度要求。文库采用BGI T7平台进行测序,测序模式为PE 150。
-
为了确保后续分析的质量,使用Stacks(V2.68)里面的process_radtags模块对下机数据进行数据质控。基于对拆分后的样品数据量统计。对单样品R1端数据,使用ustacks模块对单样品数据进行cluster聚类以及去重,形成locus。重要参数选择如下:M(1个杂合样本中2个等位基因容许的mismatch数)设置为5;m(形成聚类所需要的深度值)设置为2。对所有72个个体获得的loci位点,使用cstacks模块进行catalog文件的生成,此步参数n=5。使用sstacks模块对单个样品的SNP,allele以及tags信息比对上catalog文件,生成matches文件。运行tsv2bam以及gstacks模块进行SNP calling前的格式转换准备。运行populations模块进行共有SNP位点的calling,参数设置如下:r(该位点在单个群体的所有个体中的最低比例)为0.7,p(该位点至少需要在几个群体中存在)为3,min_maf(过滤过低频率位点)选择默认值。
-
利用Plink(V2.0)软件提取VCF中的SNP数据,然后利用Genalex(V6.51)进行遗传多样性参数计算分析。统计分析包括观察杂合性(Ho)、期望杂合性(He)和近亲繁殖指数(Fis)、遗传分化指数(Fst)等指标。此外,为评估遗传距离与地理距离之间是否存在隔离-by-距离(IBD)模式,本研究基于Fst /(1–Fst)构建了遗传距离矩阵,并使用3个样地的GPS坐标计算欧氏地理距离矩阵。随后通过R包vegan中的mantel函数进行Mantel检验。
根据高质量的SNP数据集,利用群体结构分析软件Structure(V2.3.4)进行种群遗传结构分析,其原理是基于贝叶斯聚类方法对每个样本的来源进行判断,进而反应群体的遗传结构。本次分析中,Structure利用全部位点,burn-in设置100 000,MCMC设置500 000。K设置1~15,每个K值重复5次。利用structure harverster [17−20]统计,确定最佳K值。最后,使用R语言的ggplot2包绘制种群结构分析图。
通过系统发育与主成分分析(PCA)评估3个种群之间的关系。利用poppr R软件包[21]对所有个体位点进行PCA聚类分析。
-
对来自B、M、J 3个群体的总共72个样品进行分离和过滤,共得到72.0 Gb的有效数据(干净数据),单样品平均测序量为1.0 Gb。所有样品一共获得1 257 930个catalog位点。经过SNP调用和筛选,共鉴定出8 394个高质量共有位点,每个位点上选择第1个SNP进行遗传多样性和其他分析。
-
根据SNP数据的群体遗传分析结果,海南钻喙兰3个种群(B、M、J)均表现出中等偏低的遗传多样性(表1)。其中,核苷酸多样性(Pi)为0.137–0.147,B群体的多样性最高(Pi = 0.147),M与J群体略低(Pi = 0.139和0.137)。期望杂合度(He)为0.132~0.145,而观测杂合度(Ho)普遍偏低(0.105~0.122),导致轻度至中度的近交系数(Fis = 0.049~0.107),表明群体内存在一定程度的杂合子缺失。整体样本(BWL)显示更高的近交程度(Fis = 0.219),提示存在亚结构或Wahlund效应[22]。
表 1 海南钻喙兰三个种群的遗传多样性
Table 1. Genetic diversity of three populations of Rhynchostylis gigantea.
Pop N Pi Ho He Fis BWL 72 0.149 0.116 0.149 0.219 B 46 0.147 0.122 0.145 0.107 M 11 0.139 0.121 0.132 0.049 J 15 0.137 0.105 0.132 0.104 注:Pop.种群;BWL.霸王岭样地;B.B群;M.M群;J.J群;N.样本数量;Pi.核苷酸多样性;Ho.观察杂合度;He.期望杂合度;Fis.近交系数。多样性指标均基于最终筛选后的SNP位点计算。 Note: Pop. Population; BWL.Bawangling plot; B. Population B; M. Population M; J. Population J; N. Sample size; Pi. Nucleotide diversity; Ho. Observed heterozygosity; He. Expected heterozygosity; Fis. Inbreeding coefficient. All diversity indices were calculated based on the final set of filtered SNP loci. 群体间遗传分化值(Fst)显示,B群体与M、J群体之间的Fst分别为0.028和0.026(P < 0.01),表明两者之间存在较低至中等程度的分化;而M群体与J群体之间的遗传分化值最高(Fst = 0.059,P < 0.01),显示两者间的遗传差异相对更大。总体上,3个群体间的遗传分化值处于中等水平,说明不同风水林中的群体已形成一定程度的遗传独立性,但仍保持有限的基因交流。Mantel检验结果显示遗传距离与地理距离之间不存在显著相关性(Mantel r =–
0.3086 , P = 0.667)。这表明在本研究所覆盖的小尺度空间范围内,3个海南钻喙兰群体并未呈现明显的隔离-by-距离(IBD)模式。 -
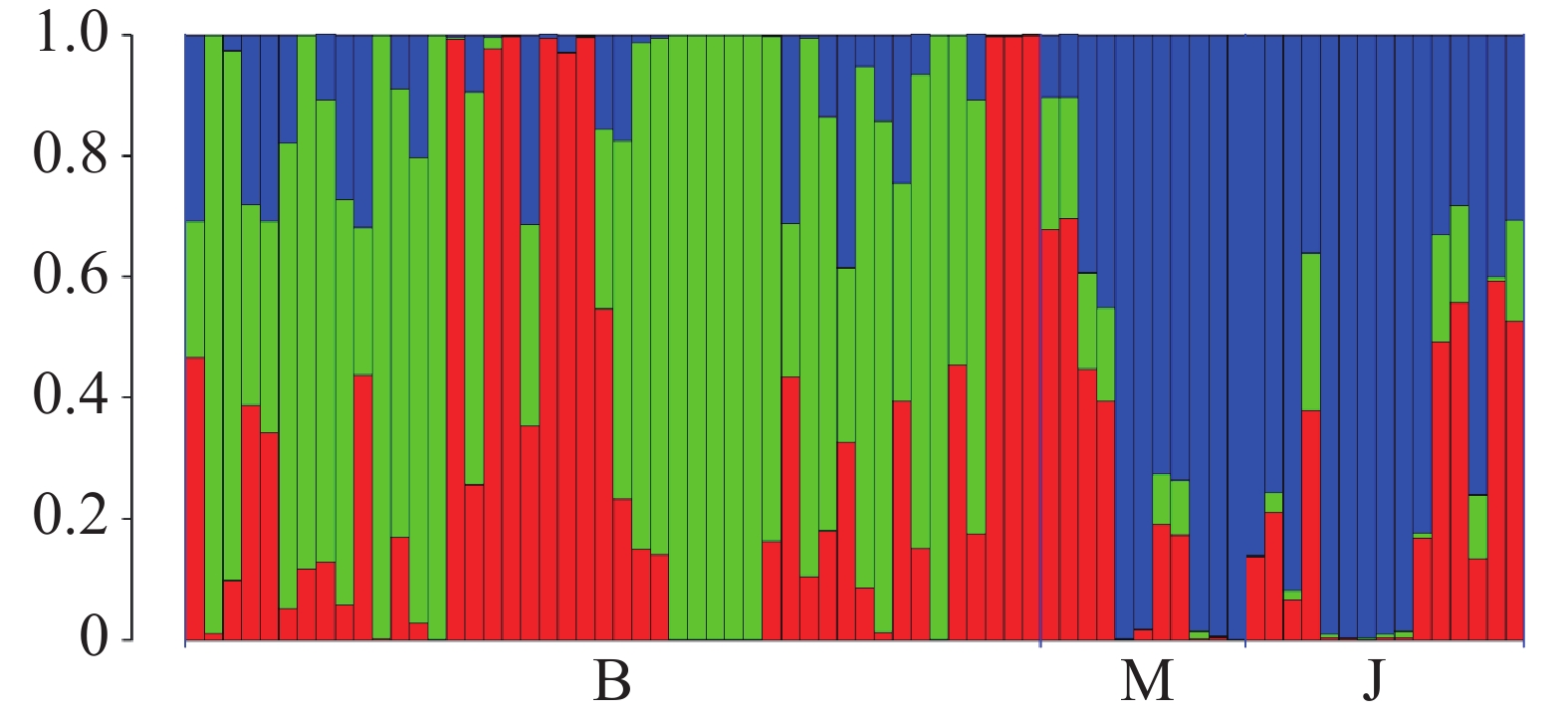
海南钻喙兰的Structure分析结果显示,delta K 在K = 3时取得最大值,故最优K值为3,即祖先种群由3种遗传成分组成。种群B的个体表现出较高程度的遗传混合,主要由绿色和红色成分组成,表明群体间存在较强的基因交流。种群M以红色成分为主,少量混入绿色和蓝色成分,显示其遗传结构相对独立但仍存在一定的基因流。相比之下,群体J几乎完全由蓝色成分构成,代表其具有独立的遗传背景,与其他群体间的基因交流较少。总体而言,这些结果表明研究群体间存在显著的遗传结构,其中群体J的遗传独立性最强。
-
主成分分析展示了海南钻喙兰3个群体(B、M、J)之间显著的遗传结构差异(图2)。PC1和PC2分别解释了4.42%和3.85%的遗传变异。尽管单轴解释率不高,但两个主成分联合足以揭示3群体的整体遗传分布格局(图2)。在PCA空间中,B群体(蓝色)样本沿PC1水平方向呈宽范围分布,形成一个显著拉长的水平椭圆,说明其个体间遗传变异较大、遗传结构更为复杂,且其分布跨越从负值到正值的多个区域。该特征与STRUCTURE分析中B群体表现出的遗传成分混合高度一致。J群体(绿色)样本主要分布于PC1负值区间,并沿PC2上下方向呈现较宽的垂直分布,意味着该群体内部存在一定遗传异质性,同时其与B群体在空间上存在部分重叠。尽管PCA显示J与B在坐标上接近,但Fst结果表明,两者仍存在中等程度的遗传分化。M群体(红色)样本在PC1和PC2坐标中均更为集中,形成陡峭、狭长的对角分布椭圆,其主轴方向与J群体近似,但两者在坐标空间中并未完全重叠,体现出一定的遗传差异。尽管M与J在PCA空间中相邻,但其Fst值为三对群体中最高,说明其等位基因频率差异较大。
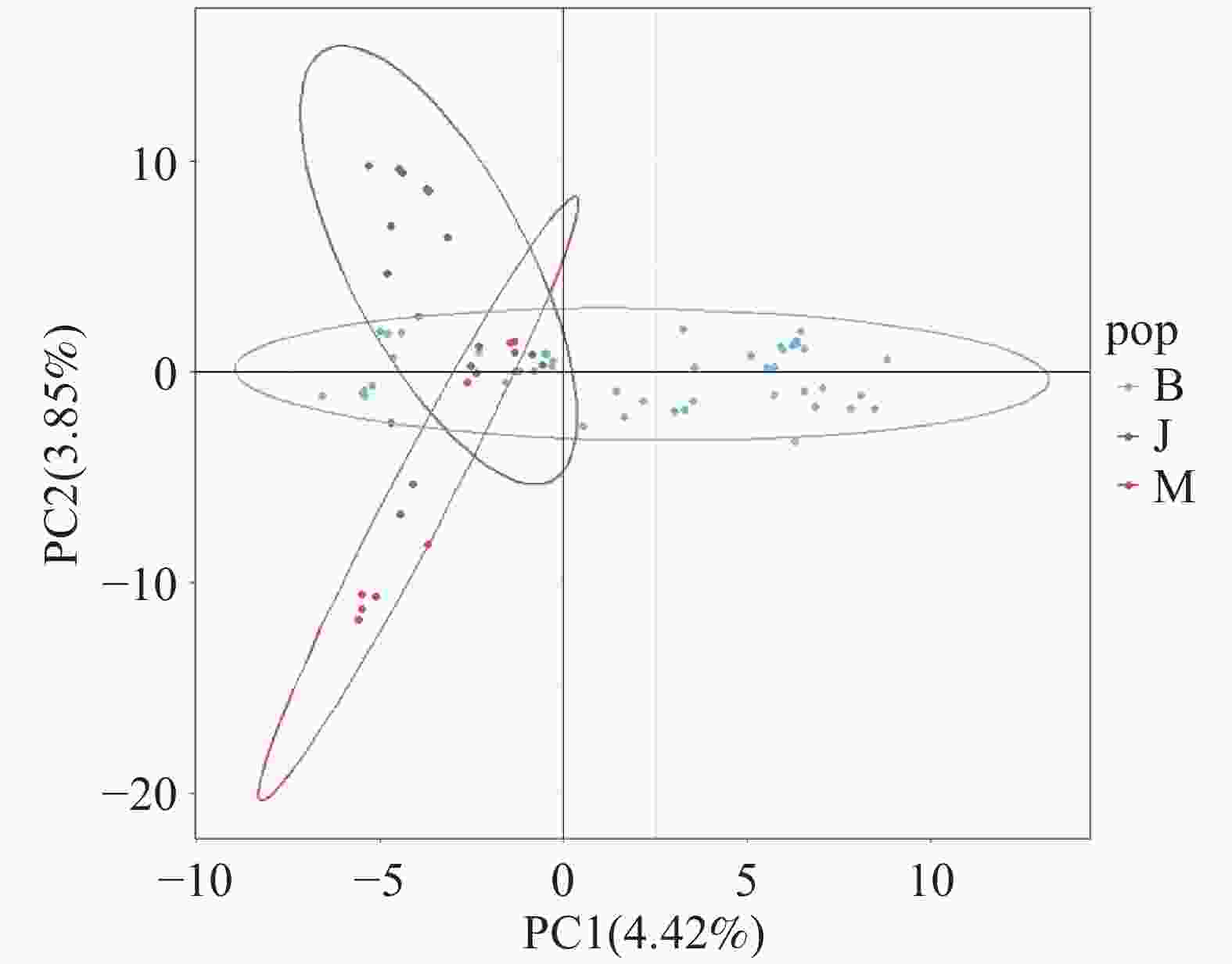
图 2 海南钻喙兰3个种群的主成分分析图
Figure 2. Principal component analysis(PCA)of the three populations of Rhynchostylis gigantea
-
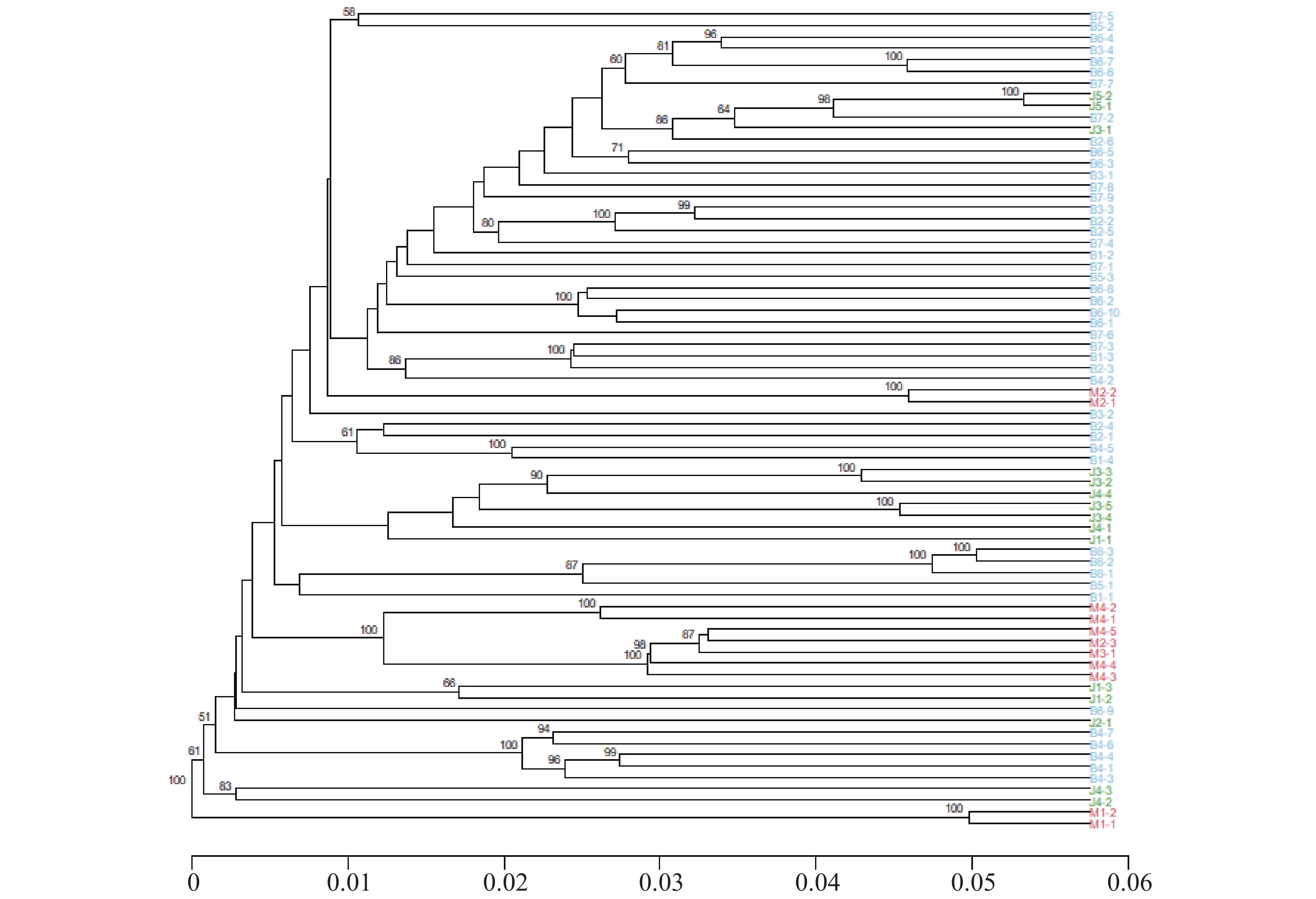
基于遗传距离的UPGMA系统发育树显示,海南钻喙兰3个群体中,群体B个体聚集紧密,形成多个高支持度(>90)的子分支,表明群体内部遗传关系密切(图3)。群体M形成独立的分支,与B群体存在中等遗传距离,而群体J位于最外侧分支,遗传距离最大(0.05~0.06),显示出较强的遗传分化与可能的长期隔离。整体聚类格局与STRUCTURE和PCA结果一致,进一步支持了3个海南钻喙兰群体中等的遗传分化与有限的基因流。
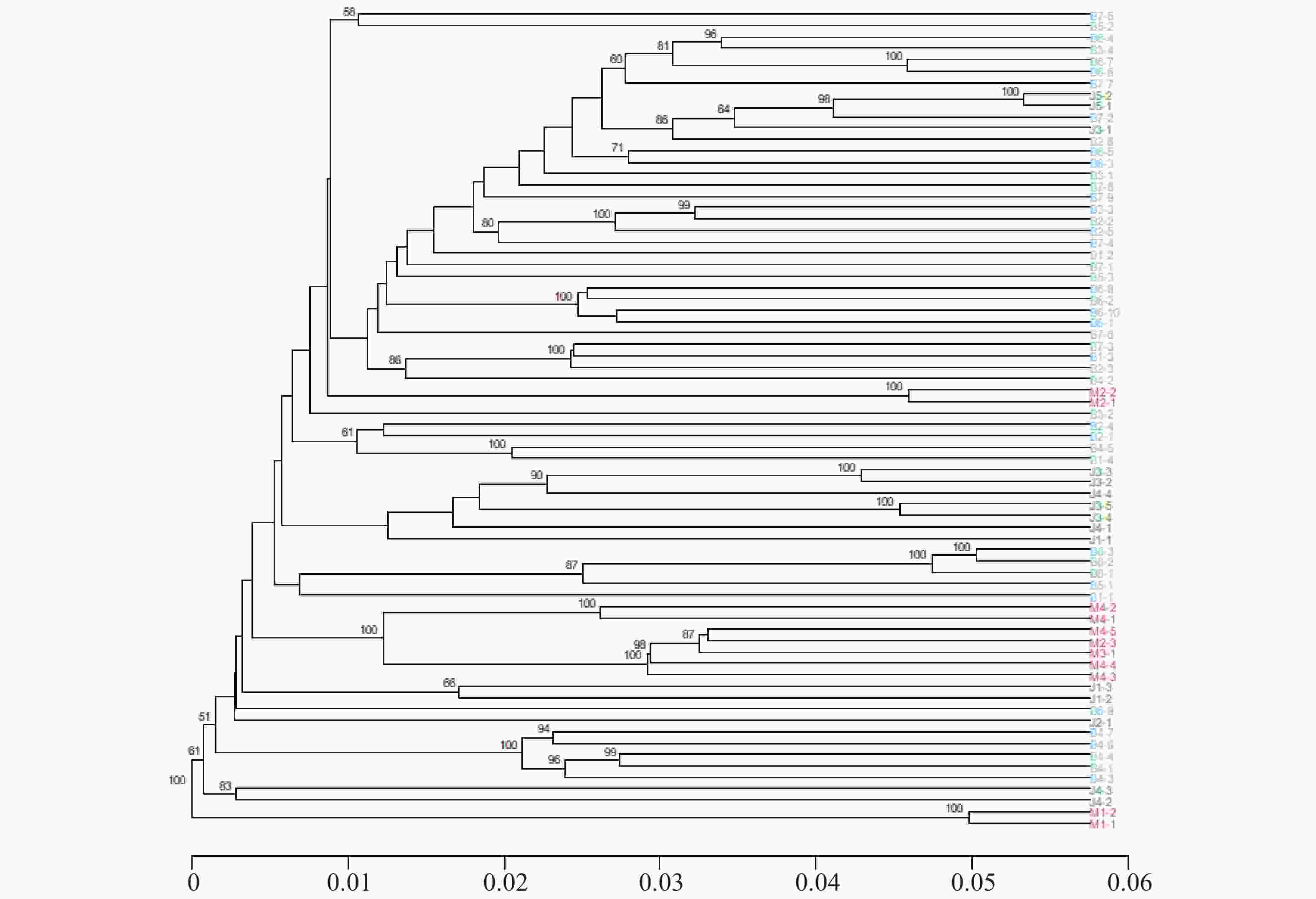
图 3 海南钻喙兰3个种群的UPGMA系统发育树
Figure 3. UPGMA phylogenetic tree of the three populations of Rhynchostylis gigantea
-
一个物种的遗传多样性高低决定了其对环境变化的适应能力和进化潜力[23−24]。一般而言,遗传多样性高的种群更有可能在多变的环境中生存。本研究通过遗传多样性参数以及遗传结构等分析,揭示了3个位于人居环境下的海南钻喙兰种群的遗传特征。结果表明,各种群的遗传多样性水平存在差异,所有种群的近交系数为正值,暗示出现了一定程度的近交倾向(Fis = 0.049~0.107)。群体遗传结构和遗传分化结果分析显示,3个种群之间存在中等水平的分化(Fst = 0.026~0.058),说明它们的基因库尚未完全混合。总体来看,那些分布在面积更小、隔离程度更高的种群往往多样性更低(如种群J,Ho = 0.105)。虽然3个群体的样本量存在差异(B = 46,M = 11,J = 15),这可能会在一定程度上影响遗传多样性参数(特别是Ho、He和Pi)的估计稳定性。然而,基于RAD-seq等高通量方法能够提供大量独立的SNP位点,即使在样本量相对有限的情况下,也可以获得较为稳定的群体结构和分化估计。相关研究发现,人为生境破碎化导致的栖息地丧失常使得小种群的遗传多样性最低,近交系数升高,与本研究发现一致[2]。与其他兰科植物的遗传多样性水平相比,如Phaius flavus(Pi =1.439 × 10−4,Ho =0.788,He= 0.707),海南钻喙兰总体(BWL)遗传多样性水平偏低(Pi = 0.149,Ho = 0.116,He = 0.149,表1),整体种群呈现更高的近交倾向(Fis = 0.219),提示整体种群中存在亚结构(Wahlund效应)[22]。本研究未检测到遗传距离与地理距离之间的显著相关性,说明3个群体之间的遗传分化并非由地理距离驱动,而更可能与风水林破碎化程度、宿主树数量减少及群体规模差异有关。
栖息地破碎化是决定种群遗传结构的重要因素,可通过影响种群规模、繁殖方式和环境因素来影响种群遗传结构[25]。村落风水林往往呈小块分布,周边被农田、道路等所隔离,这阻碍了兰花个体之间的迁移和杂交花粉传播,降低了群体间的基因流动[26]。种群尺寸一旦缩小,遗传漂变效应加剧,近交累积风险显著提高,进而降低了遗传多样性[27]。例如,Ellwanger等[2]发现,在城市化压力和林斑缩小程度较高的群体中,遗传多样性最为匮乏,且各群体普遍存在中度近交。由此可推测,海南钻喙兰中若某一风水林种群持续被隔离,其遗传变异将逐渐流失,种群遗传结构将更加分化。遗传结构图显示,3个群体(B、M、J)已形成明显的遗传分簇,其中,J群体主要由单一遗传成分组成,与其他群体的混合成分较少;而B与M群体之间存在部分颜色混合区,表明两者之间仍维持有限的基因交流(图1)。这一结构特征与PCA和UPGMA的聚类结果相吻合(图2,图3),均揭示J群体在遗传上处于相对独立状态,B与M群体则可能通过地理临近或共享的传粉者网络维持低水平的基因流。尽管STRUCTURE图中B群体与M、J群体的祖先成分模式存在明显差异,但Fst分析显示最高的遗传分化出现在M与J之间(Fst = 0.059),而B与另外2个群体之间的分化较低(Fst = 0.028 和 0.026)。这可能反映了B群体具有较高的混合程度,与M和J分别共享了较多等位基因,因此其成对Fst值相对较小。此外,M群体位于一片杧果林中,而 B与J群体主要分布于木棉林中。杧果林和木棉林的林冠结构、光照条件、树皮粗糙度与含水量均存在显著差异,这些都会影响附生兰的微生境稳定性、传粉者访问行为以及微尺度种子扩散模式。这种结构格局反映了破碎化栖息地中典型的“核心–边缘”遗传模式:边缘种群因隔离和个体数量有限而更易受到遗传漂变和近交积累的影响,从而出现遗传多样性下降和与核心种群分化加深的趋势[28]。这种由景观破碎化导致的群体分化不仅限制了基因流动,还可能削弱种群对环境变动的适应潜能。
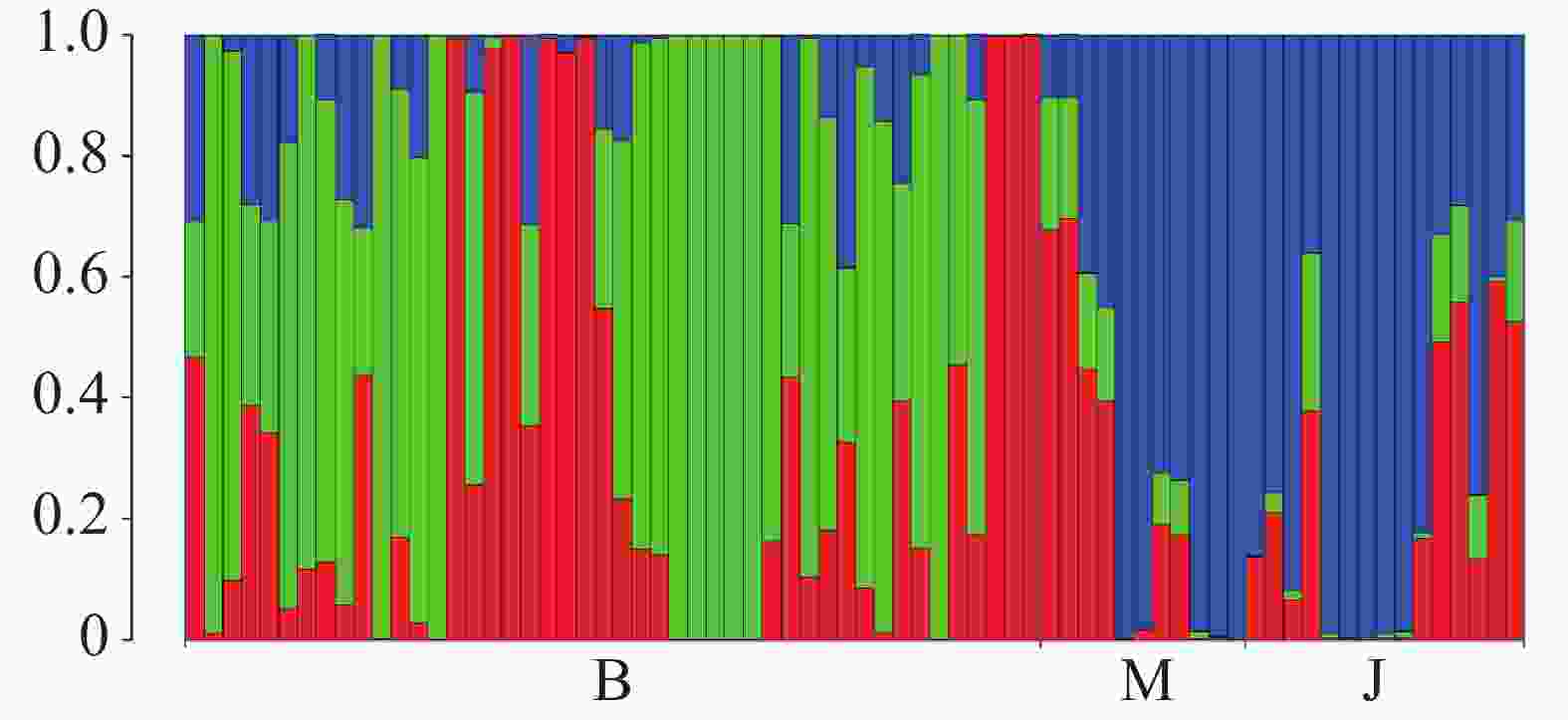
图 1 海南钻喙兰3个种群之间的遗传结构
Figure 1. Genetic structure among the three populations of Rhynchostylis gigantea
附生兰科植物对宿主树具有高度依赖性[27],其种群的存续、繁衍与扩张在很大程度上受到宿主树数量与空间分布格局的影响[22],其对环境的特殊生态需求加剧了生境破碎化的负面影响。生境破碎化不仅直接影响兰花的生长发育,还间接破坏其与传粉者、菌根真菌的共生关系,从而阻碍了种群更新能力[9]。当作为宿主树被破坏分割后,传粉昆虫种群可能因栖息地丧失而减少或消失,直接降低果实结实率[30];菌根真菌群落中断则极大抑制种子萌发,导致在后代大多在母本植株附近定植,容易引起小尺度遗传结构[31−32]。一旦种群更新率下降,兰花个体数目难以维持,自然选择和遗传漂变将进一步加剧近交和遗传分化[33]。遗传结构图显示,海南钻喙兰在人居环境中的3个种群之间仍存在一定的基因交流,这不仅归功于传粉者在花粉传播中的作用,还得益于其尘埃状微细种子能够随风远距离扩散。该特性使海南钻喙兰在一定程度上突破了林斑之间的空间隔离,维持了低水平的基因流。然而,由于种子萌发高度依赖特定的菌根真菌和适宜的微生境,这种潜在的长距离传播并不总能转化为有效的基因交流,因此在破碎化的风水林环境中,基因流仍呈受限状态。
传统遗留乔木为海南钻喙兰从原生环境传播提供了关键的生态载体与遗传通道。这些长期保留下来的古树——如木棉、杧果、枫香等——不仅在风水林文化中被视为吉祥象征而得以保存,同时生态层面上为附生兰提供了连续分布的“繁殖网络”。它们粗糙的树皮、高位分枝与常年稳定的微气候,构成了理想的附生基础,有利于花粉和种子的跨林传播与短程扩散,从而在破坏的生境中维持局部的基因流。海南钻喙兰在风水林周边区域的基因流格局,很可能依赖于这些散布的“生态岛屿”——当古树沿村道、河谷或农田边缘零星分布时,兰花个体间的花粉或种子可借助传粉者移动或干燥传播交叉完成斑块扩散,从而延缓遗传分化的加剧。然而,若这些古树因人为砍伐、更新或景观改造而减少,原有的生态性将被进一步发展,种群间基因流随之受阻,最终可能导致更为明显的遗传多样性丧失。研究调查中发现,许多木棉古树周边已被改建成槟榔种植林。随着槟榔种植的扩大,农业的用药使用显着增加,农药和除草剂的飘散不仅改变了林下微气候,也对传粉昆虫和共生真菌造成了潜在的危害,为海南钻喙兰等附生兰的生存和繁衍带来了潜在的威胁。
因此,在未来的保护与管理中,应将传统乔木视为文化与生态双重的保护单元,不仅要维持其在风水林体系中的生长,还应在风景画上规划乔木廊道或生态斑块,以增强海南钻喙兰及其他附生植物的传播与遗传联系。这种将传统乔木与生物多样性保护相结合的策略,或将成为人居环境中附生兰长期生存与续续的重要基础。
Genetic diversity of Rhynchostylis gigantea in human settlement environments
-
摘要: 以海南钻喙兰(Rhynchostylis gigantea)为研究对象,基于SNP分子标记系统分析其在破碎化风水林景观中的遗传多样性与群体结构。结果显示,3个局地群体(B、M、J)均保持中等水平的遗传多样性(Pi = 0.137~0.147,He = 0.132~0.145),但观测杂合度(Ho = 0.105~0.122)偏低,呈轻度至中度的近交系数(Fis = 0.049~0.107)。群体间的遗传分化为中等偏低(Fst = 0.026~0.058),其中,M与J群体差异最大。STRUCTURE、PCA和UPGMA分析结果一致表明,3个人居环境种群处于中等程度的遗传分化,存在一定程度的基因交流。本研究表明,即使在高度人类干扰的风水林景观中,传统遗留乔木仍能在一定程度上维持海南钻喙兰种群间的基因流,但随着宿主树减少和农业集约化增强,该类种群的遗传连通性和更新潜力可能持续下降。Abstract: A field survey of Rhynchostylis gigantea was made in fragmented fengshui forest landscapes in Changjiang, Hainan and samples of R. gigantea was collected to analyzed its genetic diversity and population structure by using SNP molecular markers. The results showed that all three local populations (B, M, and J) of R. gigantea maintained moderate levels of genetic diversity (Pi = 0.137–0.147, He = 0.132–0.145), whereas the observed heterozygosity was relatively low (Ho = 0.105–0.122), indicating mild to moderate inbreeding (Fis = 0.049–0.107). The genetic differentiation among the populations was moderate to low (Fst = 0.026–0.058), with the greatest divergence observed between the populations M and J. STRUCTURE, PCA, and UPGMA analyses consistently revealed moderate genetic differentiation among the three populations occurring in human-influenced environments, suggesting the presence of some gene flow. The field survey indicated that remnant traditional trees serve as important ecological corridors facilitating the dispersal of R. gigantea between primary and secondary habitats. However, the expansion of areca and rubber plantations surrounding these remnant trees, along with increased agrochemical use, may further weaken gene flow and population regeneration. This study highlights the genetic status of R. gigantea in fragmented landscapes and provides essential scientific insights for the conservation of its genetic resources and populations within human-influenced environments.
-
图 3 海南钻喙兰3个种群的UPGMA系统发育树
注:节点处数字表示自助法(bootstrap)支持率(%);不同颜色代表来自不同种群的个体;下方的刻度尺表示SNP遗传距离,用于量化不同个体或分支之间的遗传分化程度。
Fig. 3 UPGMA phylogenetic tree of the three populations of Rhynchostylis gigantea
Note: Numbers at the nodes indicate bootstrap support values(%). Different colors represent individuals from different populations. The scale bar at the bottom indicates SNP genetic distance, which quantifies the degree of genetic differentiation among individuals or branches.
图 1 海南钻喙兰3个种群之间的遗传结构
注:图中显示了K = 3 时的祖先系数。x轴表示3个种群中的不同个体;y轴量化了个体与推断祖先种群的变异比例。
Fig. 1 Genetic structure among the three populations of Rhynchostylis gigantea
Note: The figure shows the ancestry coefficients inferred at K = 3. The x-axis represents individual samples from the three populations, and the y-axis quantifies the proportion of genetic variation in each individual attributed to the inferred ancestral populations.
表 1 海南钻喙兰三个种群的遗传多样性
Table 1 Genetic diversity of three populations of Rhynchostylis gigantea.
Pop N Pi Ho He Fis BWL 72 0.149 0.116 0.149 0.219 B 46 0.147 0.122 0.145 0.107 M 11 0.139 0.121 0.132 0.049 J 15 0.137 0.105 0.132 0.104 注:Pop.种群;BWL.霸王岭样地;B.B群;M.M群;J.J群;N.样本数量;Pi.核苷酸多样性;Ho.观察杂合度;He.期望杂合度;Fis.近交系数。多样性指标均基于最终筛选后的SNP位点计算。 Note: Pop. Population; BWL.Bawangling plot; B. Population B; M. Population M; J. Population J; N. Sample size; Pi. Nucleotide diversity; Ho. Observed heterozygosity; He. Expected heterozygosity; Fis. Inbreeding coefficient. All diversity indices were calculated based on the final set of filtered SNP loci.  下载: 导出CSV
下载: 导出CSV
-
[1] Lienert J. Habitat fragmentation effects on fitness of plant populations–a review [J]. Journal for Nature Conservation, 2004, 12(1): 53−72. https://doi.org/10.1016/j.jnc.2003.07.002 doi: 10.1016/j.jnc.2003.07.002 [2] Ellwanger C, Steger L, Pollack C, et al. Anthropogenic fragmentation increases risk of genetic decline in the threatened orchid Platanthera leucophaea [J]. Ecology and Evolution, 2022, 12(2): e8578. https://doi.org/10.1002/ece3.8578 doi: 10.1002/ece3.8578 [3] 文亚峰, 韩文军, 吴顺. 植物遗传多样性及其影响因素[J]. 中南林业科技大学学报, 2010, 30(12): 80−87. https://doi.org/10.3969/j.issn.1673-923X.2010.12.016 doi: 10.3969/j.issn.1673-923X.2010.12.016 [4] Kramer A T, Ison J L, Ashley M V, et al. The paradox of forest fragmentation genetics [J]. Conservation Biology, 2008, 22(4): 878−885. https://doi.org/10.1111/j.1523-1739.2008.00944.x doi: 10.1111/j.1523-1739.2008.00944.x [5] Wallace L E, Bowles M L. Floral and genetic divergence across environmental gradients is moderated by inter-population gene flow in Platanthera dilatata (Orchidaceae) [J]. Frontiers in Ecology and Evolution, 2023, 11: 1085938. https://doi.org/10.3389/fevo.2023.1085938 doi: 10.3389/fevo.2023.1085938 [6] Tremblay R L, Ackerman J D, Zimmerman J K, et al. Variation in sexual reproduction in orchids and its evolutionary consequences: a spasmodic journey to diversification [J]. Biological Journal of the Linnean Society, 2005, 84(1): 1−54. https://doi.org/10.1111/j.1095-8312.2004.00400.x doi: 10.1111/j.1095-8312.2004.00400.x [7] Mccormick M K, Jacquemyn H. What constrains the distribution of orchid populations? [J]. New Phytologist, 2014, 202(2): 392−400. https://doi.org/10.1111/nph.12639 doi: 10.1111/nph.12639 [8] 葛常理. 罗氏石斛遗传多样性分析及其非共生萌发[D]. 福州: 福建师范大学, 2023. https://doi.org/10.27019/d.cnki.gfjsu.2023.000672 [9] Aguilar R, Quesada M, Ashworth L, et al. Genetic consequences of habitat fragmentation in plant populations: susceptible signals in plant traits and methodological approaches [J]. Molecular Ecology, 2008, 17(24): 5177−5188. https://doi.org/10.1111/j.1365-294X.2008.03971.x doi: 10.1111/j.1365-294X.2008.03971.x [10] 杨蕾. 大花杓兰适生区预测、种群遗传多样性及基因流研究[D]. 北京: 北京林业大学, 2022. https://doi.org/10.26949/d.cnki.gblyu.2022.001310 [11] Lei J R, Chen Y Q, Li L M, et al. Spatiotemporal change of habitat quality in Hainan Island of China based on changes in land use [J]. Ecological Indicators, 2022, 145: 109707. https://doi.org/10.1016/j.ecolind.2022.109707 doi: 10.1016/j.ecolind.2022.109707 [12] 颜平, 黄明忠, 杨光穗, 等. 海南钻喙兰种质资源现状调查[J]. 广东农业科学, 2015, 42(5): 24−30. https://doi.org/10.16768/j.issn.1004-874x.2015.05.021 doi: 10.16768/j.issn.1004-874x.2015.05.021 [13] 陈馷嶂, 赵莹, 张哲, 等. 海南钻喙兰开花物候与繁殖特性研究[J]. 植物科学学报, 2025, 43(4): 444−453. https://doi.org/10.11913/PSJ.2095-0837.24194 doi: 10.11913/PSJ.2095-0837.24194 [14] 帖聪晓. 基于简化基因组测序RAD-seq的寒兰景观遗传学研究[D]. 南昌: 南昌大学, 2022. https://doi.org/10.27232/d.cnki.gnchu.2022.002223 [15] 刘财国. 基于SNP的福建武夷山和建瓯茶树种质资源遗传多样性分析[D]. 福州: 福建农林大学, 2023. https://doi.org/10.27018/d.cnki.gfjnu.2023.000186 [16] Doyle J J, Doyle J L. Isolation of plant DNA from fresh tissue [J]. Focus, 1990, 12(1): 13−15. [17] Pritchard J K, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data [J]. Genetics, 2000, 155(2): 945−959. https://doi.org/10.1093/genetics/155.2.945 doi: 10.1093/genetics/155.2.945 [18] Falush D, Stephens M, Pritchard J K. Inference of population structure using multilocus genotype data: dominant markers and null alleles [J]. Molecular Ecology Notes, 2007, 7(4): 574−578. https://doi.org/10.1111/j.1471-8286.2007.01758.x doi: 10.1111/j.1471-8286.2007.01758.x [19] Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study [J]. Molecular Ecology, 2005, 14(8): 2611−2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x doi: 10.1111/j.1365-294X.2005.02553.x [20] Earl D A, Vonholdt B M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method [J]. Conservation Genetics Resources, 2012, 4(2): 359−361. https://doi.org/10.1007/s12686-011-9548-7 doi: 10.1007/s12686-011-9548-7 [21] Kamvar Z N, Tabima J F, Grünwald N J. Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction [J]. PeerJ, 2014, 2: e281. https://doi.org/10.7717/peerj.281 doi: 10.7717/peerj.281 [22] De Meeûs T. Revisiting FIS, FST, Wahlund effects, and null alleles [J]. Journal of Heredity, 2018, 109(4): 446−456. https://doi.org/10.1093/jhered/esx106 doi: 10.1093/jhered/esx106 [23] Frankham R. Genetic rescue of small inbred populations: meta-analysis reveals large and consistent benefits of gene flow [J]. Molecular Ecology, 2015, 24(11): 2610−2618. https://doi.org/10.1111/mec.13139 doi: 10.1111/mec.13139 [24] Hedrick P W. Recent developments in conservation genetics [J]. Forest Ecology and Management, 2004, 197(1/3): 3−19. https://doi.org/10.1016/j.foreco.2004.05.002 doi: 10.1016/j.foreco.2004.05.002 [25] Liang C Y, Li J, Li S X, et al. Human activity changed the genetic pattern of the orchid Phaius flavus population [J]. Diversity, 2024, 16(11): 685. https://doi.org/10.3390/d16110685 doi: 10.3390/d16110685 [26] Ellstrand N C. Is gene flow the most important evolutionary force in plants? [J]. American Journal of Botany, 2014, 101(5): 737−753. https://doi.org/10.3732/ajb.1400024 doi: 10.3732/ajb.1400024 [27] Cascante-Marín A, Oostermeijer G, Wolf J, et al. Genetic diversity and spatial genetic structure of an epiphytic bromeliad in Costa Rican montane secondary forest patches [J]. Biotropica, 2014, 46(4): 425−432. https://doi.org/10.1111/btp.12119 doi: 10.1111/btp.12119 [28] Parab G V, Krishnan S. Assessment of genetic variation among populations of Rhynchostylis retusa, an epiphytic orchid from Goa, India using ISSR and RAPD markers [J]. Indian Journal of Biotechnology, 2008, 7(3): 313−319. [29] De L C, Biswas S S. Adaptational mechanisms of epiphytic orchids: a review [J]. International Journal of Bio-resource and Stress Management, 2022, 13(11): 1312−1322. https://doi.org/10.23910/1.2022.3115a doi: 10.23910/1.2022.3115a [30] Abeli T, Jäkäläniemi A, Wannas L, et al. Pollen limitation and fruiting failure related to canopy closure in Calypso bulbosa (Orchidaceae), a northern food-deceptive orchid with a single flower [J]. Botanical Journal of the Linnean Society, 2013, 171(4): 744−750. https://doi.org/10.1111/boj.12014 doi: 10.1111/boj.12014 [31] Jacquemyn H, Brys R, Vandepitte K, et al. Fine‐scale genetic structure of life history stages in the food‐deceptive orchid Orchis purpurea [J]. Molecular Ecology, 2006, 15(10): 2801−2808. https://doi.org/10.1111/j.1365-294X.2006.02978.x doi: 10.1111/j.1365-294X.2006.02978.x [32] Sletvold N, Joffard N, Söderquist L. Fine-scale genetic structure in the orchid Gymnadenia conopsea is not associated with local density of flowering plants [J]. American Journal of Botany, 2024, 111(2): e16273. https://doi.org/10.1002/ajb2.16273 doi: 10.1002/ajb2.16273 [33] Tremblay R L, Ackerman J D. Gene flow and effective population size in Lepanthes (Orchidaceae): a case for genetic drift [J]. Biological Journal of the Linnean Society, 2001, 72(1): 47−62. https://doi.org/10.1111/j.1095-8312.2001.tb01300.x doi: 10.1111/j.1095-8312.2001.tb01300.x -
 点击查看大图
点击查看大图
计量
- 文章访问数: 448
- HTML全文浏览量: 212
- 被引次数: 0
