

-
蜱虫(Ixodida: Ixodidae)作为仅次于蚊子的第二大节肢动物病原传播媒介,对全球公共卫生、家畜健康及野生动物种群构成了严重威胁[1]。蜱虫能够携带并传播多种病原体,包括蜱传脑炎病毒(TBEV)[2−3]、立克次体(Rickttsia spp)、伯氏疏螺旋体(Borrelia burgdorferi)、巴贝西虫(Babesia spp)、巴尔通体(Bartonella spp)、嗜吞噬细胞无形体(Anaplasma phagocytophilum)、土拉弗朗西斯菌(Francisella tularensis)和柯克斯体(Coxiella spp)等多种人畜共患病[1]。全球已知蜱虫种类约840余种[4],而在中国,已记录的种类超过120种[5],包含110多种硬蜱,11种软蜱[6]。中国国内蜱虫携带的微生物种类丰富,尤其细菌类多样性极高,且不同地区、蜱种携带的微生物存在显著差异[7]。
传统上,对蜱传病原体的研究主要依赖于分离培养和基于PCR的靶向检测,这些方法虽然有效,但存在检测病原种类局限、无法发现未知病原体以及难以揭示微生物群落整体互作网络的局限性[1]。随着高通量测序技术的飞速发展,宏基因组学(Metagenomics)成为蜱虫微生物组研究的核心手段。2024年,曹务春团队通过宏基因组学对中国48种蜱虫的蜱传微生物的组成进行了分析,为蜱虫病的防控和诊断工具的开发提供了新的视角[7]。
海南岛气候属于热带季风海洋性气候,全年温暖湿润的气候条件为蜱虫的生存和繁衍提供了适宜的生态生存环境。蜱虫通过叮咬吸血和附着寄生,传播多种病原,对人类健康构成威胁,并对动物的生产性能与整体健康造成损害,对畜牧业与宠物行业造成显著的经济损失[8]。蜱虫体内的微生物组成受环境、宿主等因素影响[7],然而,现阶段针对海南岛生态环境中蜱虫微生物的组成与功能仍缺乏系统研究。
埃里希体属是蜱传的一类具有重要公共卫生意义的病原体,其中,米氏埃里希体(Ehrlichia minasensis)于加拿大和巴西等地有广泛报道,在2019年之后陆续在中国境内(海南、昆明等地)的动物体内有该病原体的报道[33−34]。米氏埃里希体与犬埃里希体(Ehrlichia canis)的全基因组高度同源[21],均能导致发热及血液系统异常,与犬埃里希体的症状高度相似,但米氏埃里希体比犬埃里希体的宿主更广泛,在牛、羊等反刍动物上均有检测到[35]。米氏埃里希体虽目前尚无感染人的报道,但作为埃里希体属成员,其传播媒介蜱虫可同时携带多种对人致病的埃里希体属病原体,因此,在蜱虫活跃的海南对埃里希体属开展研究,对评估和防范其公共卫生风险具有重要意义。
本研究核心目标在于利用宏基因组学手段研究海南岛部分地区蜱虫微生物组的组成结构及其功能,同时针对性研究埃里希体属在蜱虫中的组成情况。这不仅有助于推动新型防控策略的制定,还可为特异性诊断靶标的发掘提供关键数据基础,从而支持蜱传疾病的前期预警与精准诊断[7]。
-
本研究中使用的主要实验仪器包括:连续变倍体视显微镜(型号 XTL-BM-7B,上海彼爱姆光学仪器制造有限公司)、移液器(型号 Eppendorf N13462C,Eppendorf)、小型离心机(型号 ABSON MiFly-6,合肥艾本森科学仪器有限公司;型号 Eppendorf
5430 R,Eppendorf)、高速台式冷冻离心机(型号 Eppendorf 5424R,Eppendorf)、超微量分光光度计(型号 NanoDrop2000,Thermo Fisher Scientific)、酶标仪(型号 BioTek ELx800,BioteK)、旋涡混合器(型号 QL-901,海门其林贝尔仪器制造有限公司)、粉碎研磨仪(型号 TL-48R,上海万柏生物科技有限公司)、MP 研磨仪(型号 FastPrep-24 5G,MP)、微型荧光计(型号 Quantus™ Fluorometer,Promega)、磁力架(生工生物工程(上海)股份有限公司)、电泳仪(型号 DYY-6C,北京市六一仪器厂)、PCR 仪(型号 ABI GeneAmp® 9700 型,ABI)及测序仪(型号 Illumina MiSeq®,Illumina)。使用的主要实验试剂包括:Blood-DNA Extraction kit、2×TaqPlus Master-Mix II(Dye Plus)(均由南京诺维赞生物科技股份有限公司提供),DL2000-DNA Marker、DL500-DNA Marker(均由北京宝日医生物技术有限公司提供),FastDNA® Spin Kit for Soil(MP Biomedicals,美国),Biowest agarose(Biowest,西班牙),以及 Pure red 核酸染料(兰杰柯科技有限公司)。 -
蜱虫采集:在海南中部定安县新竹镇、南部万宁市兴隆华侨农场进行犬只身上捕捉蜱虫;在海南西部白沙黎族自治县打安镇可程村五指山猪科技小院内的五指山猪上捕捉蜱虫。蜱虫样品信息及分组情况具体见表1。其中,A组蜱虫取自流浪犬,在定安县新竹镇流浪动物中心群居圈养;B组蜱虫取自白沙特色猪种五指山猪身上,采用“圈地散养”的模式饲养在山上;C组蜱虫取自万宁本地村民散养家养犬。对同一乡镇不同的犬只(或猪只)身上收集到的蜱虫按采集宿主分类,并分为吸血状态和未吸血状态,在采集管上标注。收集的蜱虫保存在95%酒精中,置于-20℃环境保存。随后在实验室进行分组,从3个地区未吸血的蜱虫组中各取出3只,将定安犬源蜱虫定义为A组,3只分别标记为A1、A2和A3;白沙猪源的蜱虫定义为B组,分别标记为B1、B2、B3;万宁犬源的蜱虫定义为C组,3只分别标记为C1、C2和C3。组内各样本来自非同一宿主。
表 1 蜱虫样品信息及分组情况
Table 1. Sample Information and Grouping Details of Ticks
样本分组
Sample group样本名称
Sample name样本来源
Sample source采集宿主
Collection host采集状态
Collection status采集时间
Collection timeA A1 定安县新竹镇
Xinzhu Town, Ding'an犬
Canis未吸血,雌蜱
unfed female ticks2024-09-24 A2 A3 B B1 白沙黎族自治县打安镇可程村五指猪科技小院
Wuzhishan Breed Science and Technology
Courtyard, Kecheng Village, Da'an Town,
Baisha Li Autonomous County猪
Sus未吸血,雌蜱
unfed female ticks2025-01-11 B2 B3 C C1 万宁市兴隆镇华侨农场
Xinglong Overseas Chinese Farm,
Xinglong Town, Wanning City犬
Canis未吸血,雌蜱
unfed female ticks2025-04-09 C2 C3 -
形态学鉴定:将各组蜱虫通过体视显微镜观察形态,参照《中国重要医学昆虫分类与鉴别》[9]和《常见医学蜱螨图谱》[10]进行形态学鉴定。
PCR方法鉴定:使用液氮研磨粉碎,使用DNA提取试剂盒(南京诺唯赞公司)提取DNA,以DNA为模板分别对16s、Nad5基因PCR扩增,引物见表2,使用1%琼脂糖凝胶电泳,胶回收后送至生工生物工程(上海)股份有限公司进行测序,最终确定蜱虫的种类。
表 2 血红扇头蜱和长角血蜱PCR引物序列
Table 2. PCR primer Sequences of Rhipicephalus sanguineus and Haemaphysalis longicornis
引物名称
Primer name引物序列(5'—3')
Primer sequence(5'—3')扩增长度/bp
Amplicon length /bp物种
Species参考文献
ReferencesRs-16S-F CTGCTCAATGATTTTTTAAATTGCTGTGG 460 Rhipicephalus sanguineus [11] Rs-16S-R CCGGTCTGAACTCAGATCAAGT 460 Rhipicephalus sanguineus [11] Nad-5-F TCTAAAATTAAATCCTTTGAAT 500 Haemaphysalis longicornis [12] Nad-5-R AAGAGCCCAAATTCCATTTTC -
将蜱虫样品送至武汉爱基百客生物科技有限公司,分别对3组蜱虫采用95%乙醇及ddH2O进行清洗,去除表面的微生物。清洗过后的蜱虫置于超净台中风干水分,使用FastDNA® Spin Kit for Soil(MP Biomedicals)提取试剂盒提取总DNA,超微量紫外可见分光光度计检测DNA纯度及浓度,1%琼脂糖凝胶电泳检测DNA完整性。将质检合格的DNA构建PE文库,使用DNBSEQ-T7进行PE150测序。
原始测序数据使用fastp(v0.20.0)进行质控过滤,具体参数为在50 bp的滑动窗口内,若平均质量值低于20则截去后端序列;剔除长度不足50 bp的reads以及包含N碱基的reads。随后,利用FLASH(v1.2.7)对双端reads进行拼接,最小重叠区为10 bp,重叠区允许的最大错配率为0.2。使用UPARSE软件(v7.1)在97%的相似度水平下进行OTU聚类。物种分类注释通过RDP classifier(v2.2)与Silva 16S rRNA数据库(v138)完成,置信度阈值设定为70%。最后,通过KEGG数据库比对,获得基因功能注释信息。
-
对武汉爱基百客生物科技有限公司宏基因测序后返回的DNA扩增埃里希体16s rRNA基因和TRP36基因并测序,引物见表3,使用1.5%琼脂糖凝胶电泳,胶回收后送至生工生物工程(上海)股份有限公司进行测序,最终确定所含的种类。
表 3 埃里希体属PCR引物序列
Table 3. PCR primer Sequences of the Ehrlichia Genus
引物名称
Primer name引物序列(5—3')
Primer sequence(5'—3')扩增长度 /bp
Amplicon length /bp物种
Species参考文献
ReferencesTRP36-F2 TTTAAAACAAAATTAACACACTA 891 E. minasensis [13] TRP36-R1 AAGATTAACTTAATACTCAATATTACT ECC AGAACGAACGCTGGCGGCAAGC 477 Ehrlichia Spp 16s [14] ECB CGTATTACCGCGGCTGCTGGCA ECAN5 CAATTATTTATAGCCTCTGGCTATAGGA 396 E. canis HE3 TATAGGTACCGTCATTATCTTCCCTAT HE1 CAATTGCTTATAACCTTTTGGTTATAAAT 396 E .chaffeensis HE3 TATAGGTACCGTCATTATCTTCCCTAT EE52 CGAACAATTCCTAAATAGTCTCTGAC 396 E. ewengii HE3 TATAGGTACCGTCATTATCTTCCCTAT -
白沙县猪体表的蜱虫(B组)、万宁市犬体表的蜱虫(C组)在体视显微镜下显示假头宽短呈六角形,侧角明显,基突宽短,须肢粗短,第1、2 腹面内缘具粗长刚毛且排列紧密,盾板长大于宽、后缘钝圆,新鲜虫体呈褐色带光泽,刻点粗细不一,颈沟前部深陷且呈八字形向后延伸,气门板呈逗点形,眼大呈卵圆形,足跗节腹面具小齿,爪垫较小等形态学特点(图1-A,B),参照《中国重要医学昆虫分类与鉴别》[9]和《常见医学蜱螨图谱》[10],鉴定为血红扇头蜱。定安县犬体表采集的蜱虫(A组)具有假头短宽呈矩形,侧缘平行,后缘平直,基突短小,盾板呈心形,刻点细密,颈沟深呈弧形,末端达盾板后 1/3,气门板大呈圆形,足基节Ⅰ内距发达呈锥形,跗节Ⅳ窄长和亚端部渐窄的形态学特征(图1-C, D),参照《中国重要医学昆虫分类与鉴别》[9]和《常见医学蜱螨图谱》[10],鉴定为长角血蜱。

图 1 3组蜱虫体视显微镜拍摄图
Figure 1. Stereomicroscopic images of three groups of ticks
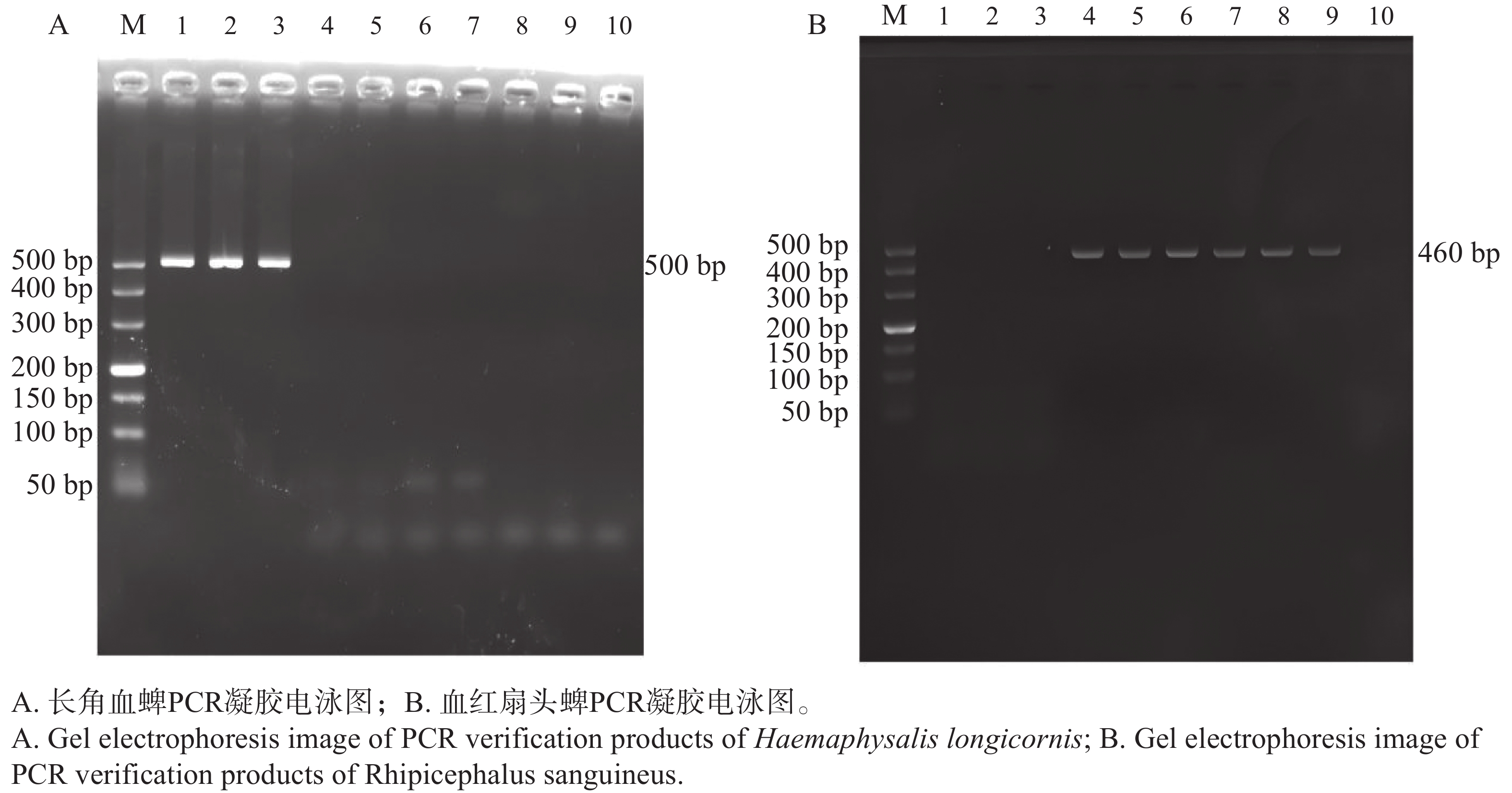
以血红扇头蜱的 16S rRNA 基因与长角血蜱的 Nad-5 基因为靶标进行 PCR 扩增后,将产物经 2% 琼脂糖凝胶电泳分离,通过凝胶成像系统获取图像(图2)。结果显示,图2 中, 16S rRNA 基因扩增片段与标准分子量Marker相比,接近稍高于500 bp 处,Nad-5基因扩增片段与标准分子量Marker相比,位于400 bp上,500 bp下,两种基因的扩增产物条带大小均与预期基因条带大小相符。将扩增产物测序后,将序列在 NCBI 数据库中进行比对,进一步确定定安犬只体表的蜱虫(A1-A3)为长角血蜱,白沙猪只体表蜱虫(B1-B3)与万宁犬只体表的蜱虫(C1-C3)为血红扇头蜱。
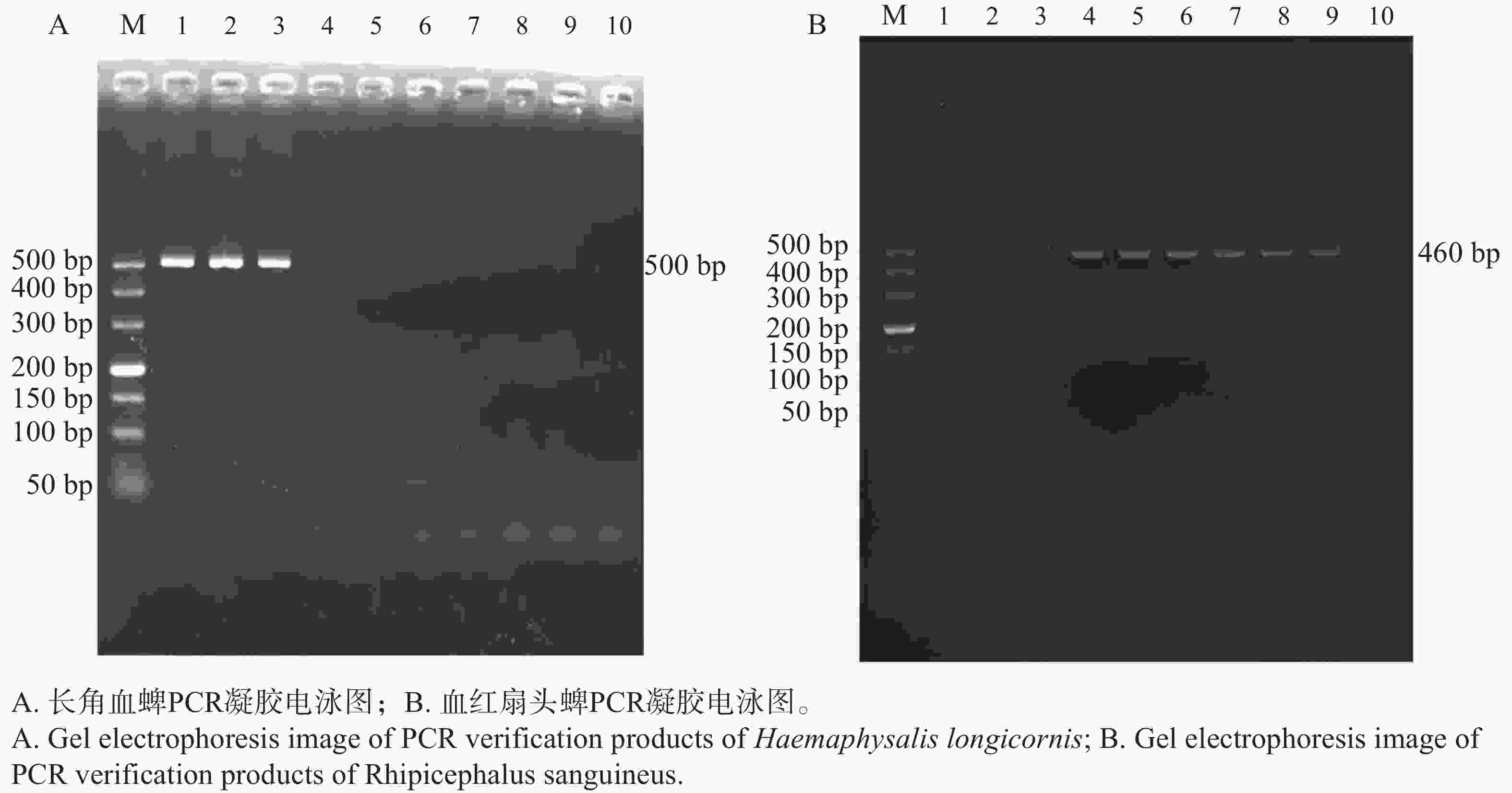
图 2 3组蜱虫PCR鉴定产物凝胶电泳图
Figure 2. Gel electrophoresis images of PCR identification products from three groups of ticks
-
3组蜱虫样本宏基因测序共得到原始数据92.91G,有效数据77.6 G,数据有效率为83.52%,GC占比48.28%,Q30占比96.29%(表4)。
表 4 海南3个不同地区采集蜱虫宏基因测序数据
Table 4. Metagenomic Sequencing Data of Ticks Collected from Three Different Regions in Hainan
样本分组
Sample group样本名称
Sample game原始数据(G)
Raw data(G)有效数据(G)
Valid data(G)有效数据率(%)
Valid data rate(%)GC(%)
GC content(%)Q30% A A1 9.87 9.01 91.17 47.65 96.39 A2 9.42 8.62 A3 10.60 9.62 B B1 10.27 9.37 90.31 50.12 95.97 B2 8.83 7.95 B3 9.48 8.50 C C1 14.94 10.67 71.18 47.08 96.51 C2 8.89 6.53 C3 10.59 7.30 合计92.89 合计77.57 注:GC占比为有效数据中两种碱基占总碱基的占比;Q30是测序数据中有效数据质量值大于或等于30的碱基所占的百分比。 Note:GC content refers to the proportion of guanine and cytosine bases relative to the total bases in the valid data; Q30 represents the percentage of bases in the valid sequencing data with a quality value greater than or equal to 30. -
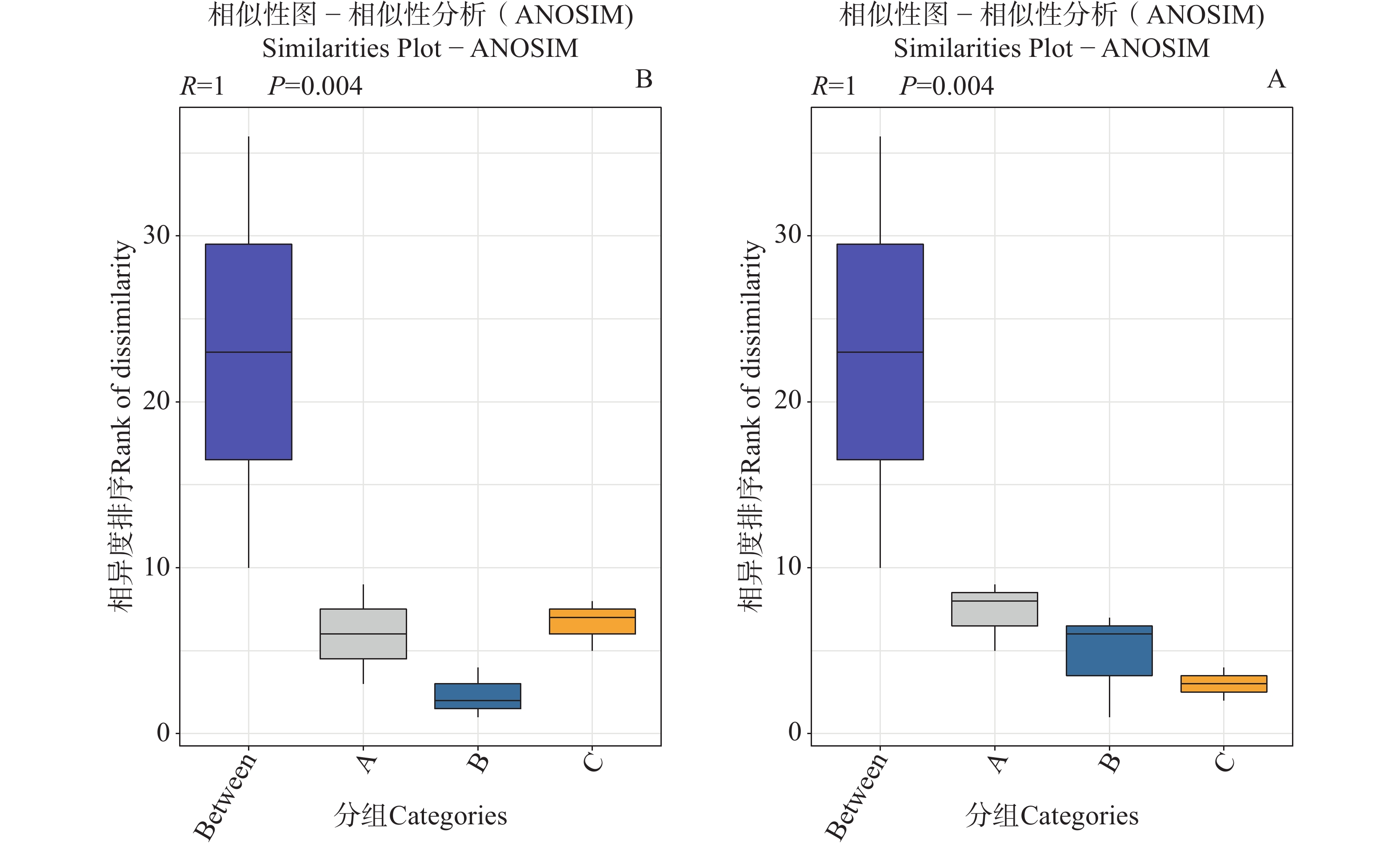
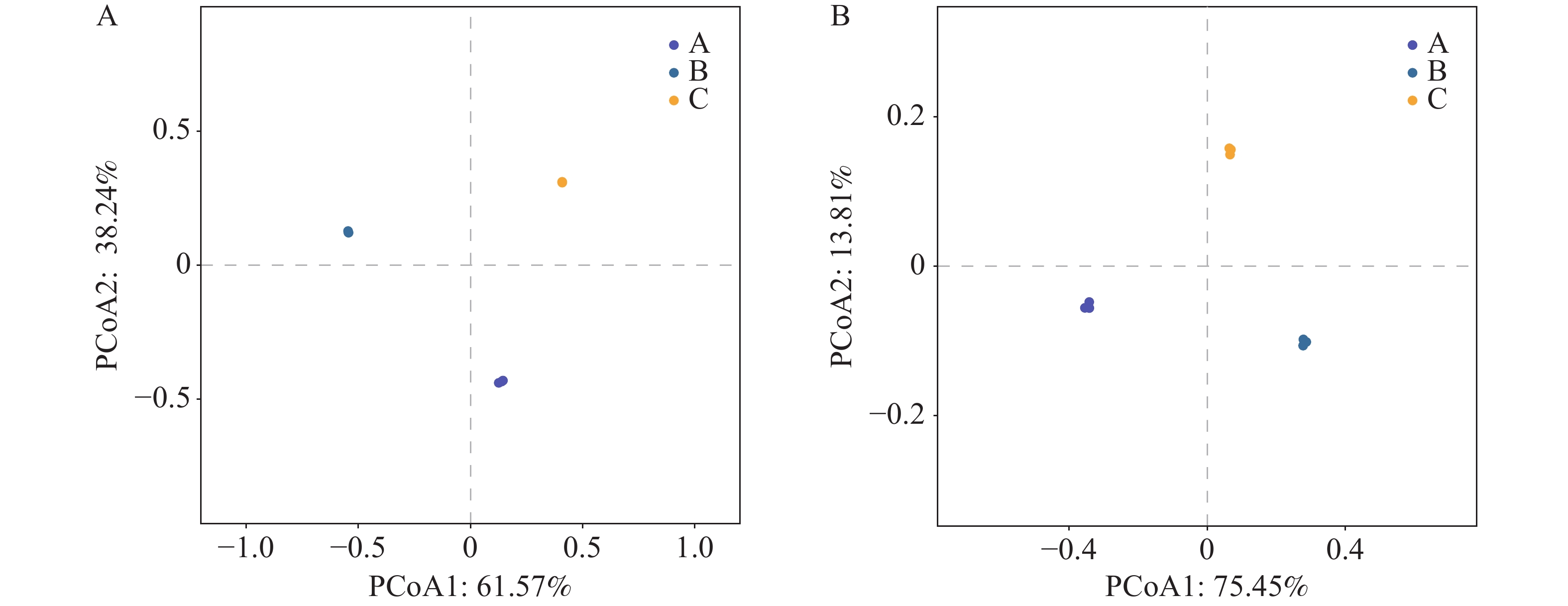
为验证组间蜱虫携带微生物群落组成的生物学差异,本研究基于门水平与种水平的物种丰度数据进行相似性分析(ANOSIM)与主坐标分析(PCoA)。ANOSIM 分析显示,全局检验及门、种水平检验均得到 R=1(P=0.004),箱线图直观显示 A、B、C 各组间的相异度秩次显著高于组内,表明组间微生物群落差异远大于组内差异,分组对群落结构具有高度区分度且该效应在不同分类水平上稳定存在。PCoA 结果显示,门水平前两轴累计贡献率达 99.81%(PCoA1: 61.57%, PCoA2: 38.24%),种水平累计贡献率达 99.39%(PCoA1: 62.77%, PCoA2: 36.62%);高累计贡献率表明前两个主坐标轴可完整反映群落结构的变异信息,且图中 A、B、C 三组样本点各自聚集,组间空间距离显著大于组内,进一步印证了组间群落结构的显著分异。上述结果表明,组间的微生物群落结构存在极显著差异,该差异主要由蜱虫种类、宿主类型及地理区域等因素共同驱动,鉴于组内异质性较低且组间差异远大于组内差异,后续分析将基于 A、B、C 三组的组间比较展开。
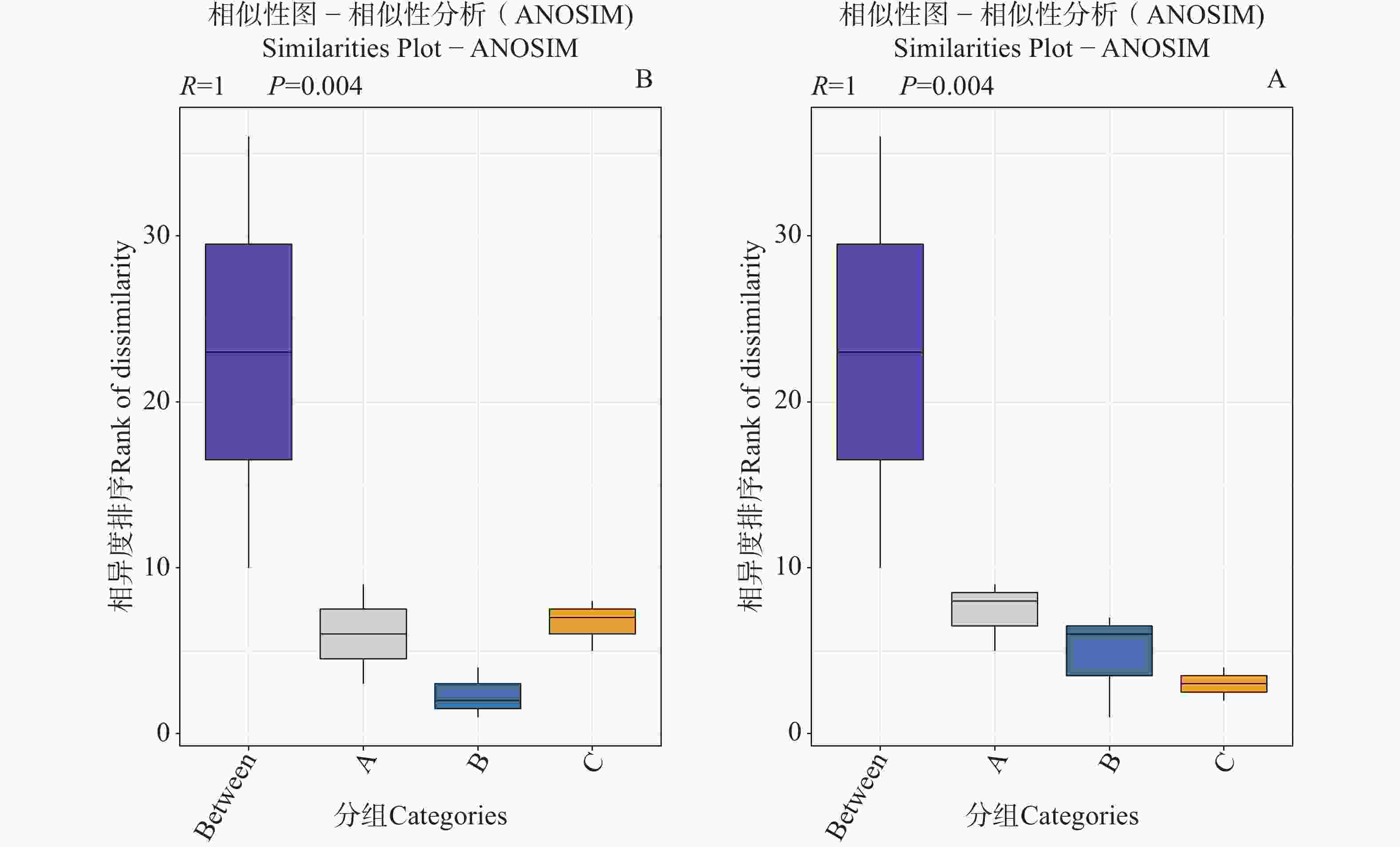
图 3 不同地区的蜱虫门水平(A)和种水平(B)物种丰度数相似性分析图
Figure 3. Species richness similarity analysis plots of ticks from different regions at the phylum level (Panel A)and species level (Panel B)
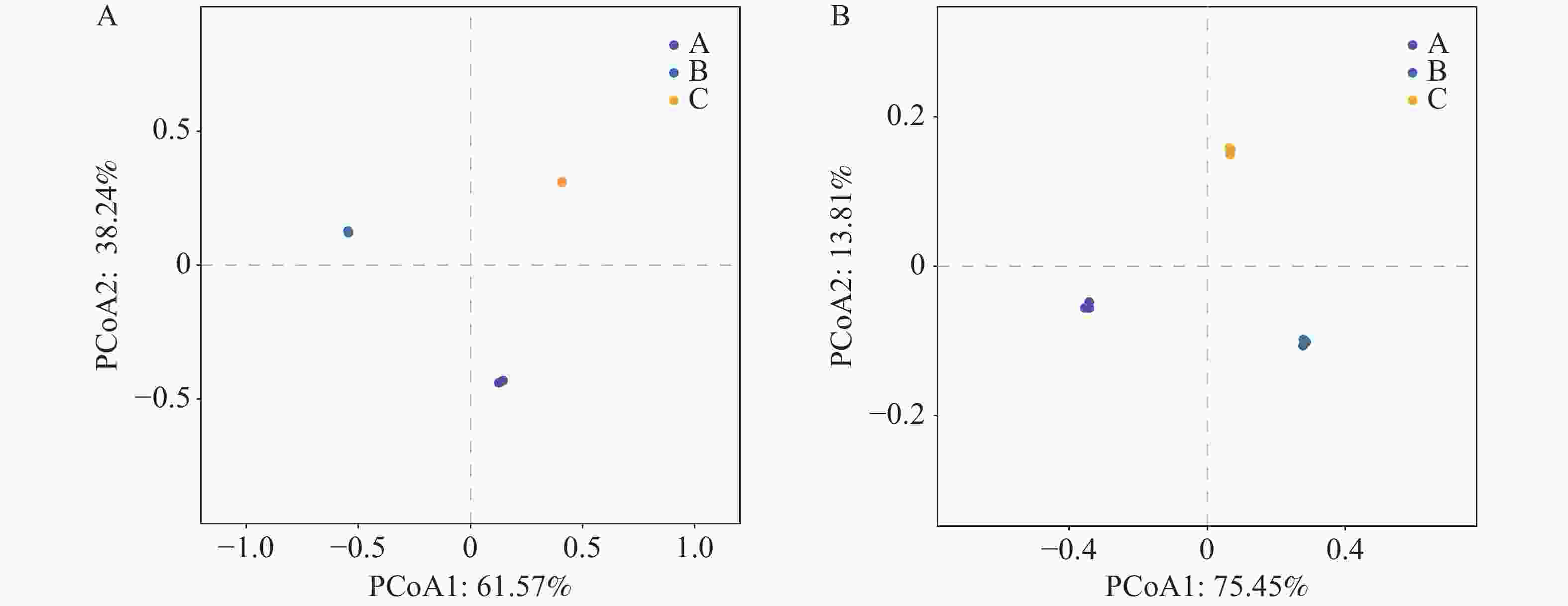
图 4 不同地区不同蜱虫门水平(A)和种水平(B)的主坐标分析
Figure 4. Principal coordinate analysis (PCoA)plots of ticks from different regions at the phylum level (Panel A)and species level (Panel B)
-
1)细菌注释结果与分析
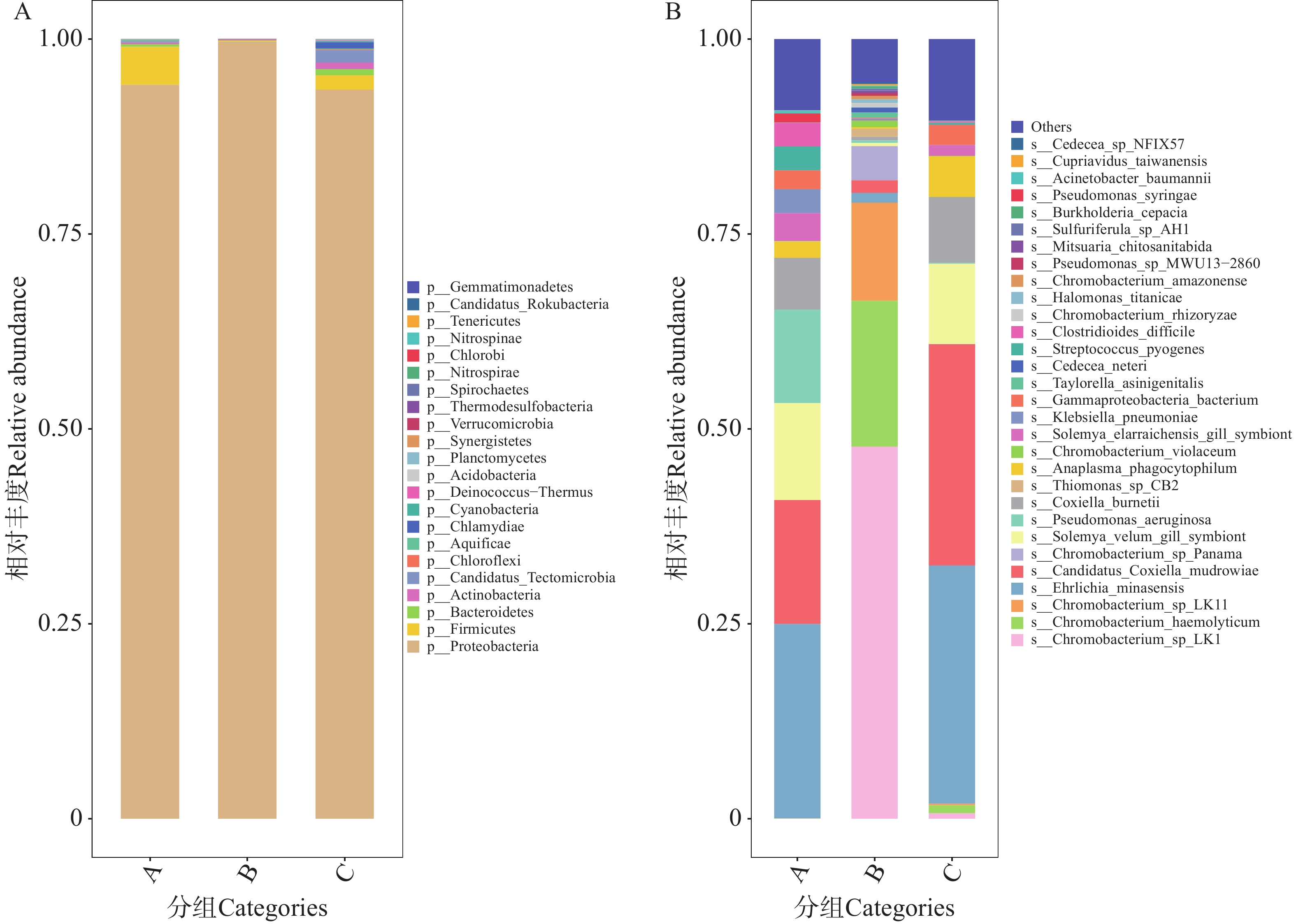
A、B、C组蜱虫携带的细菌群落在门水平均呈现单一优势门绝对主导的特征,优势门为变形菌门(Proteobacteria)。该优势门的相对丰度90%左右,而其他门[如厚壁菌门(Firmicutes)]占比极低,整体门水平多样性较低(图5-A)。共有533种细菌注释到种水平上,相对丰度排名前30的物种丰度图见图5-B。其中,能导致人兽共患病的有伯氏柯克斯体(Coxiella burnetii)、化脓性链球菌(Streptococcus pyogenes)、溶血性色杆菌(Chromobacterium haemolyticum)、嗜吞噬细胞无形体。嗜吞噬细胞无形体可导致发热和免疫系统异常;伯氏柯克斯体可导致生殖系统感染,化脓性链球菌可导致全身感染;溶血性色杆菌部分菌株可引起人和动物的感染性疾病。以上几种细菌均能对动物生产以及人类的健康造成极大的危害[17−20]。值得注意的是,本研究在海南蜱虫体内首次检测到米氏埃里希体,该病原体在海南的首次检出记录来自海南省海口市某羊场采集的羊血样本[34]。米氏埃里希体在A组、C组中呈现较高丰度,约占30%。该病原体由犬埃里希体突变而来,可引起宿主发热及血液系统异常,且其宿主范围较犬埃里希体更广(如反刍动物等),对畜牧业具有显著潜在危害[21]。其余小部分物种,例如艰难梭菌(Clostidioides difficile)、铜绿假单胞菌(Pseudomonas aeruginosa)等大多为条件致病菌,能导致人类或动物败血症、腹泻、肠炎等症状[22−23]。
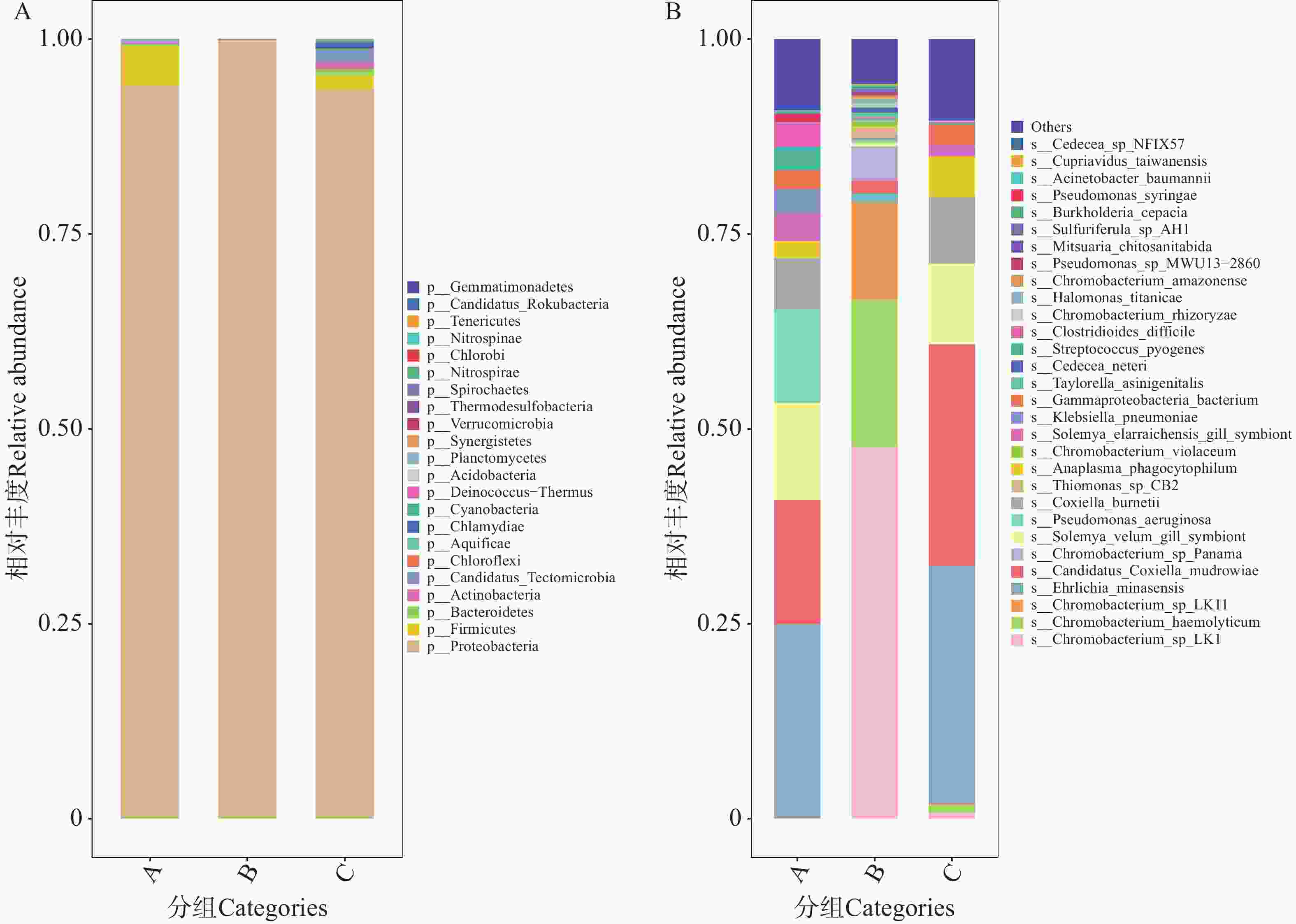
图 5 不同地区的蜱虫携带的细菌门水平(A)和种水平(B)上的相对丰度
Figure 5. Relative abundance of bacteria carried by ticks from different regions at the phylum level (Panel A)and species level (Panel B)
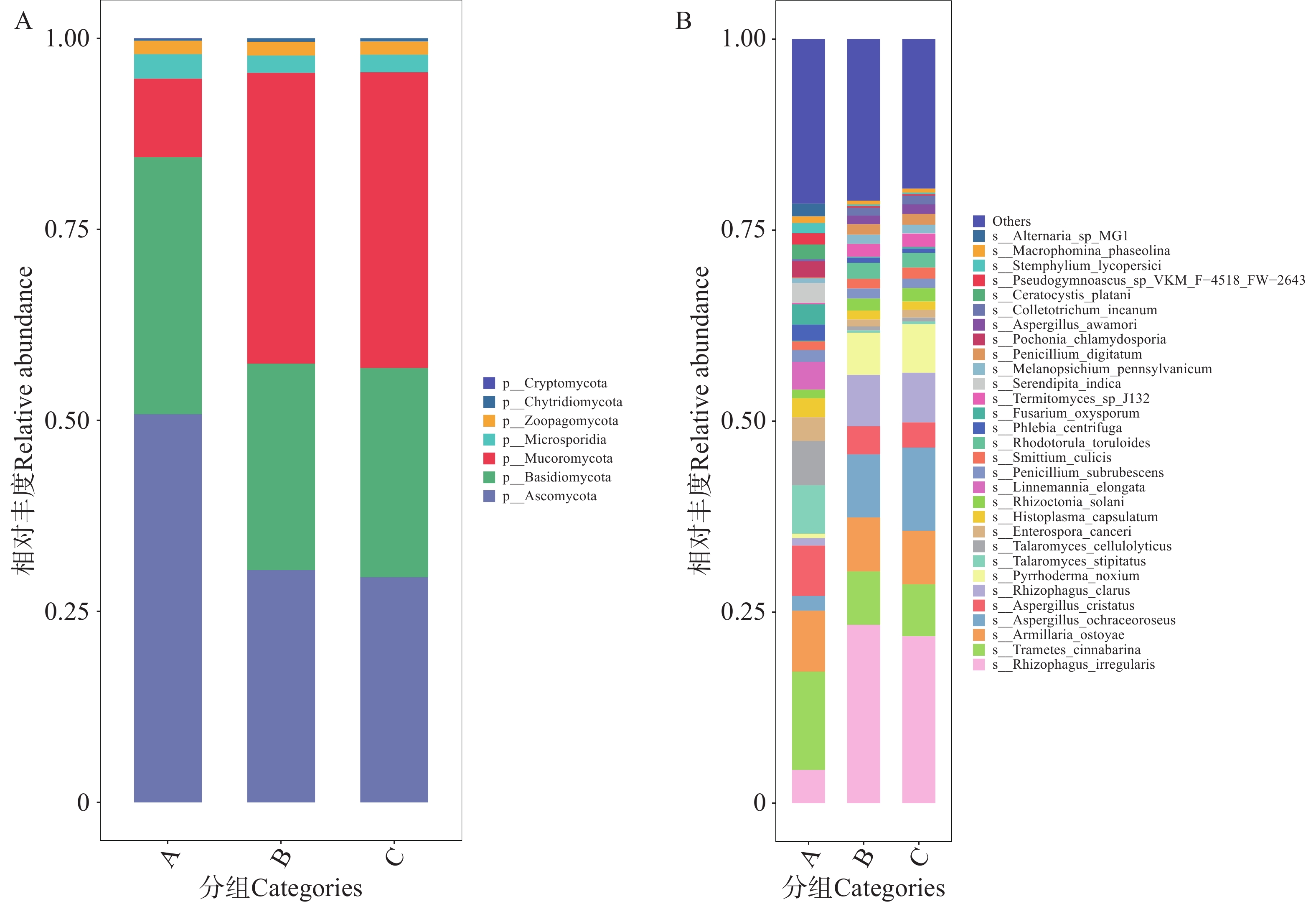
2)真菌注释结果与分析
图6-A展示了A组真菌的第一优势门为子囊菌门(Ascomycota),其相对丰度约50.79%。B、C组真菌分布具有相似度,其第一优势门为毛霉门(Mucoromycota),B、C组毛霉门的相对丰度分别为38.05%和38.71%;其第二优势门则为子囊菌门(Ascomycota),B、C组子囊菌门的相对丰度分别约为30.39%和29.37%。共有205种真菌注释到种水平上,图6-B展示了排名前30的物种。其中,10余种会对人类和动物产生危害,如赭曲霉(Aspergilluso chraceoroseus),可产赭曲霉毒素,损伤肝肾且具有致癌性,危害人类和动物健康;荚膜组织胞浆菌(Histoplasma capsulatum),可引发组织胞浆菌病,侵犯肺、骨髓等器官,对人类及免疫低下动物危害大等[24−25]。
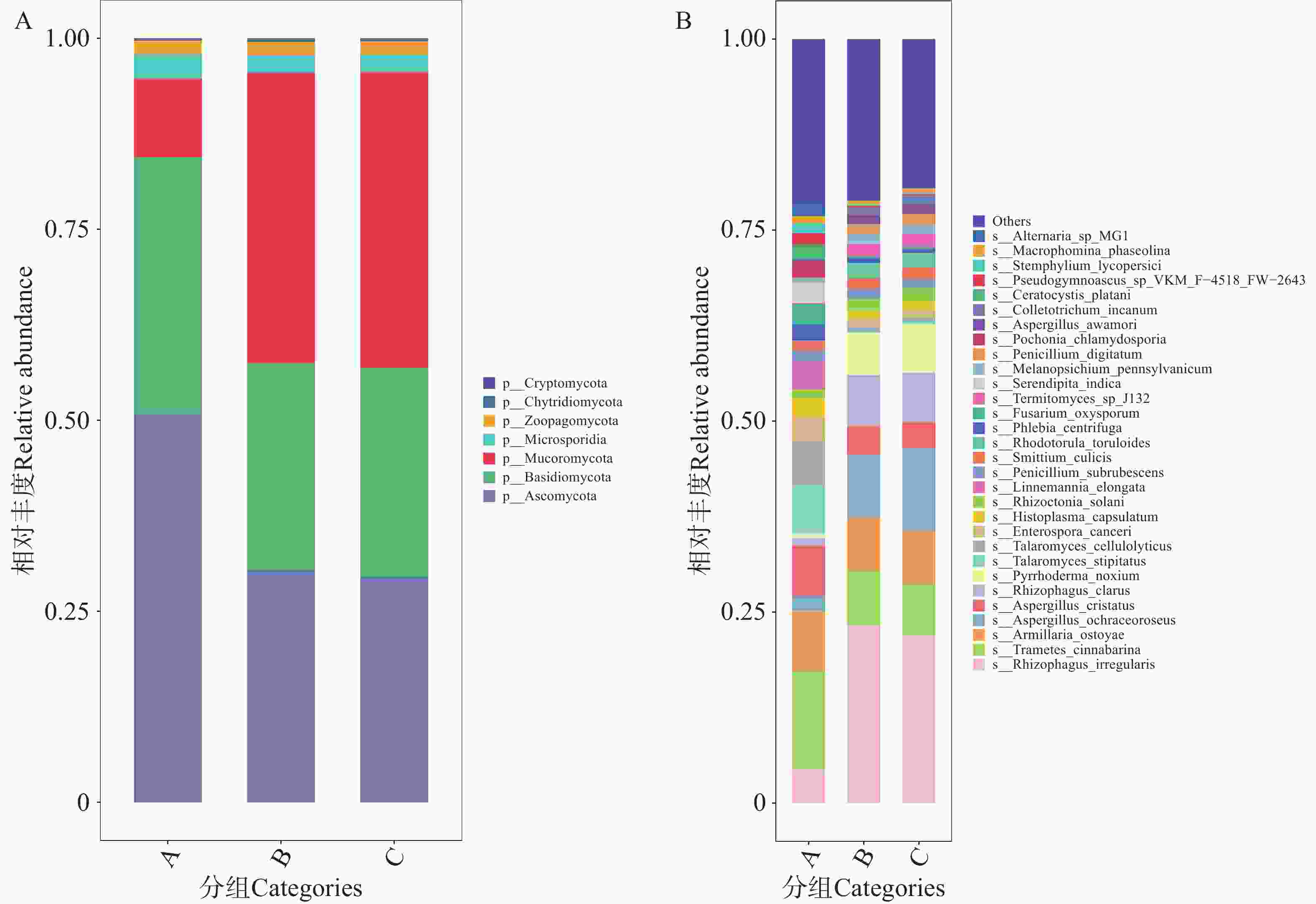
图 6 不同地区的蜱虫携带的真菌门水平(A)和种水平(B)上的相对丰度
Figure 6. Relative abundance of fungi carried by ticks from different regions at the phylum level (Panel A)and species level
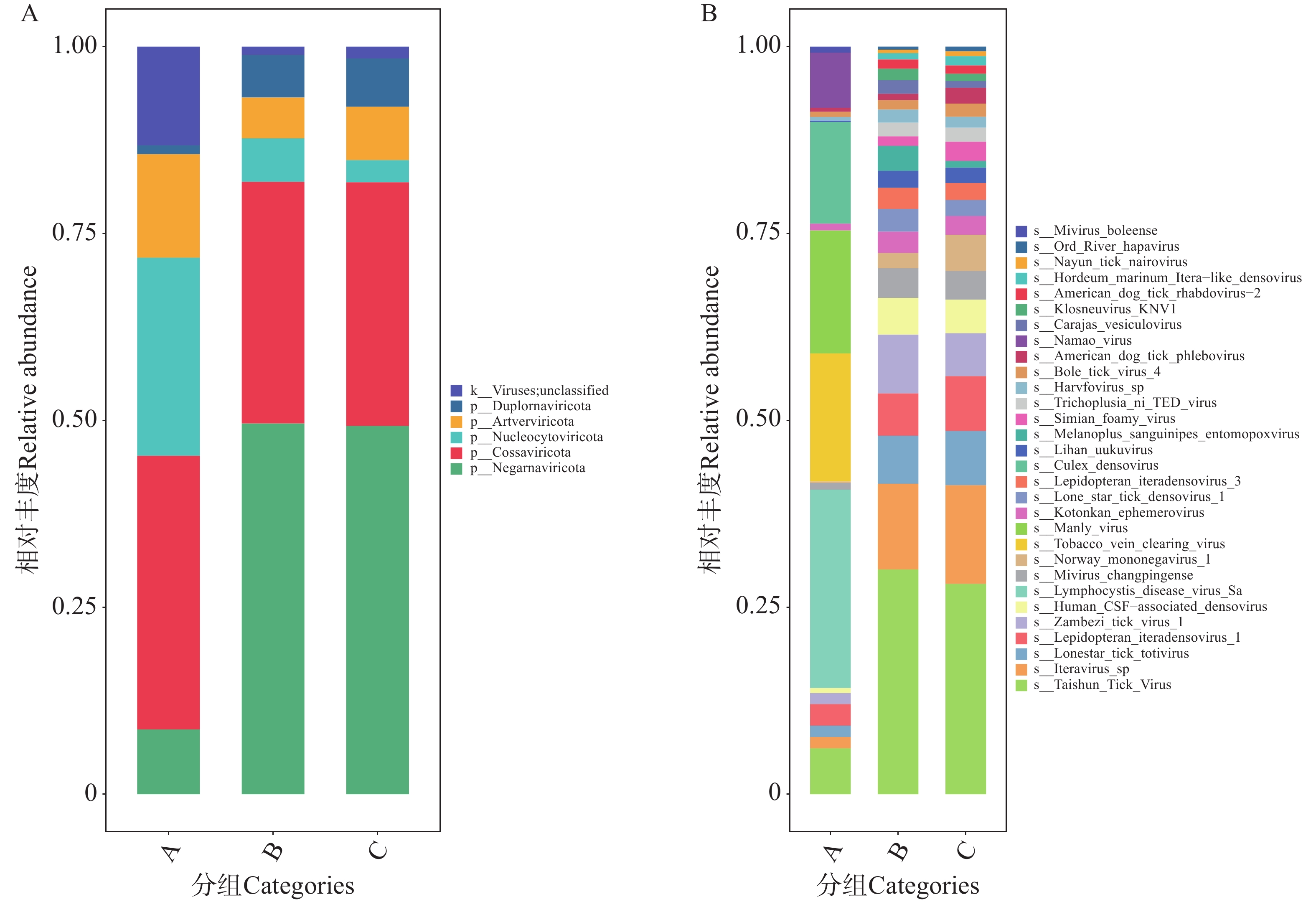
3)病毒注释结果与分析
图7-A展示了A组的优势门为和科索病毒门(Cossaviricota),而B、C组的优势门均为负链RNA病毒门(Negarnaviricota)。三组样本共注释到30种病毒(图7-B),病毒种类上,A组种类数量最少,B组与C组种类数量相当。A组病毒物种多样性最低。B、C组病毒群落组成更趋同,优势种占比显著,物种集中性更高。其中A组的优势病毒为淋巴囊肿病病毒Sa株(Lymphocystis disease virus Sa);B、C组的优势病毒为泰顺蜱病毒(Taishun Tick Virus),均占比30%左右。注释的病毒多数具有强致病性。静脉病毒(Phlebo virus),如美洲犬蜱静脉病毒(American dog tick phlebo virus),常引发发热、头痛、肌肉疼痛,部分可进展为出血热(表现为发热、皮肤黏膜出血、休克等)。弹状病毒(Rhabdo virus),如美洲犬蜱弹状病毒2型(American dog tick rhabdo virus-2)易侵犯神经系统,可出现脑炎症状(头痛、呕吐、意识障碍等),伴随发热、肌痛。浓核病毒(Denso virus),如孤星蜱浓核病毒(Lone star tick denso virus 1)多为隐性感染或轻度发热,免疫低下时可能引发全身炎症反应[26−28]。其他蜱传病毒,如泰顺蜱病毒(Taishun Tick Virus)常与发热综合征相关,表现为持续发热、乏力、血小板减少(类似“发热伴血小板减少综合征”的症状模式)[29]。
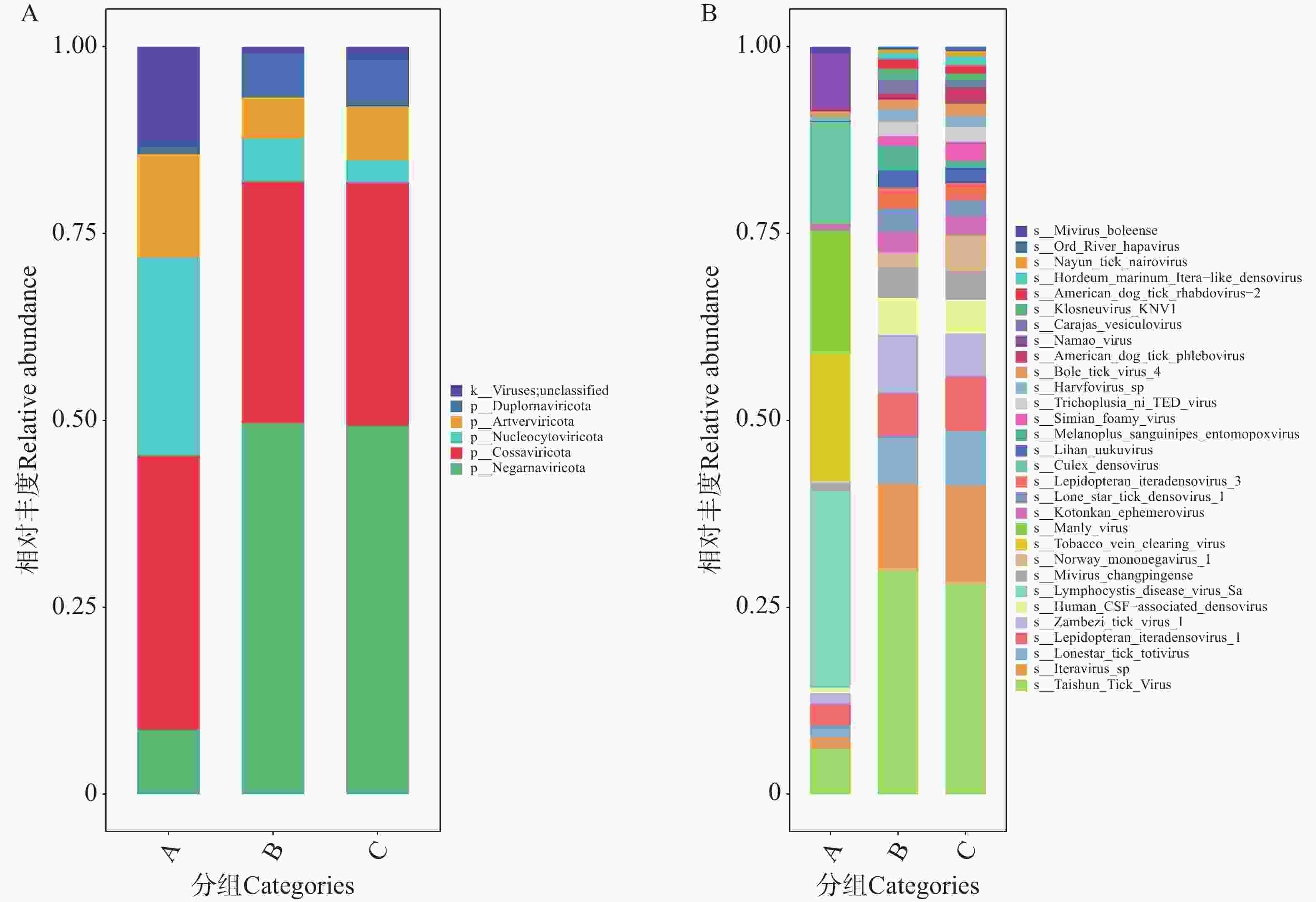
图 7 不同地区的蜱虫携带的病毒门水平(A)和种水平(B)上的相对丰度
Figure 7. Relative abundance of viruses carried by ticks from different regions at the phylum level (Panel A)and species level
-
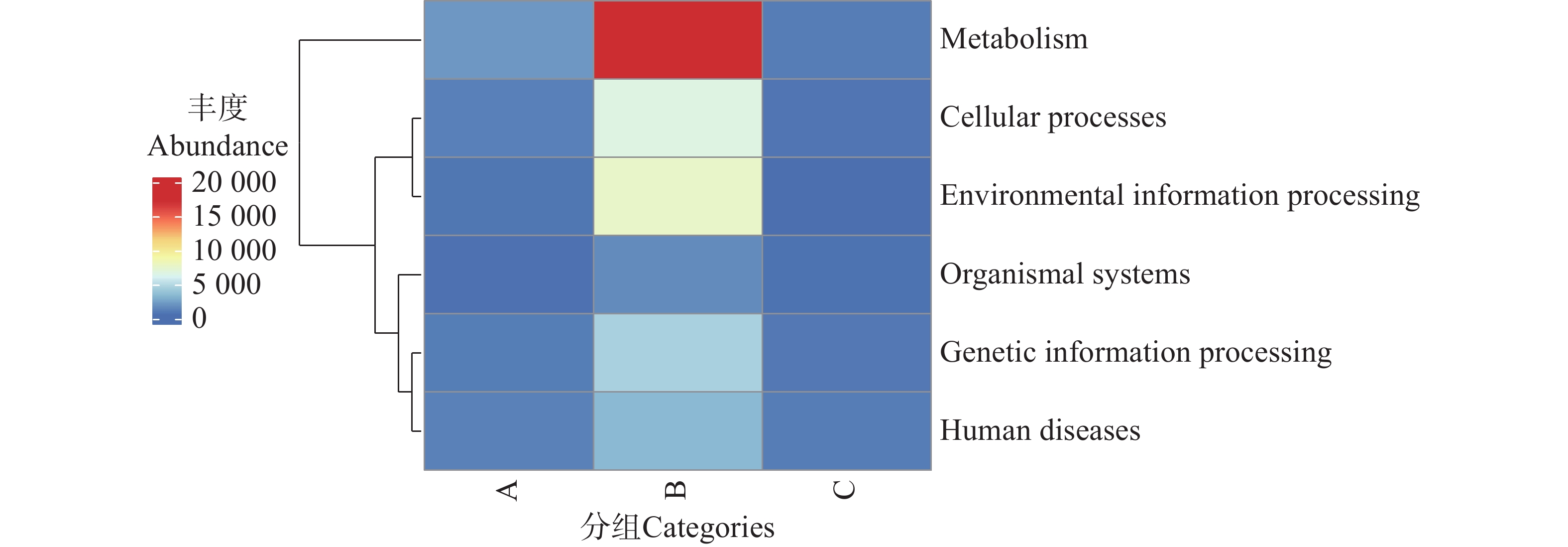
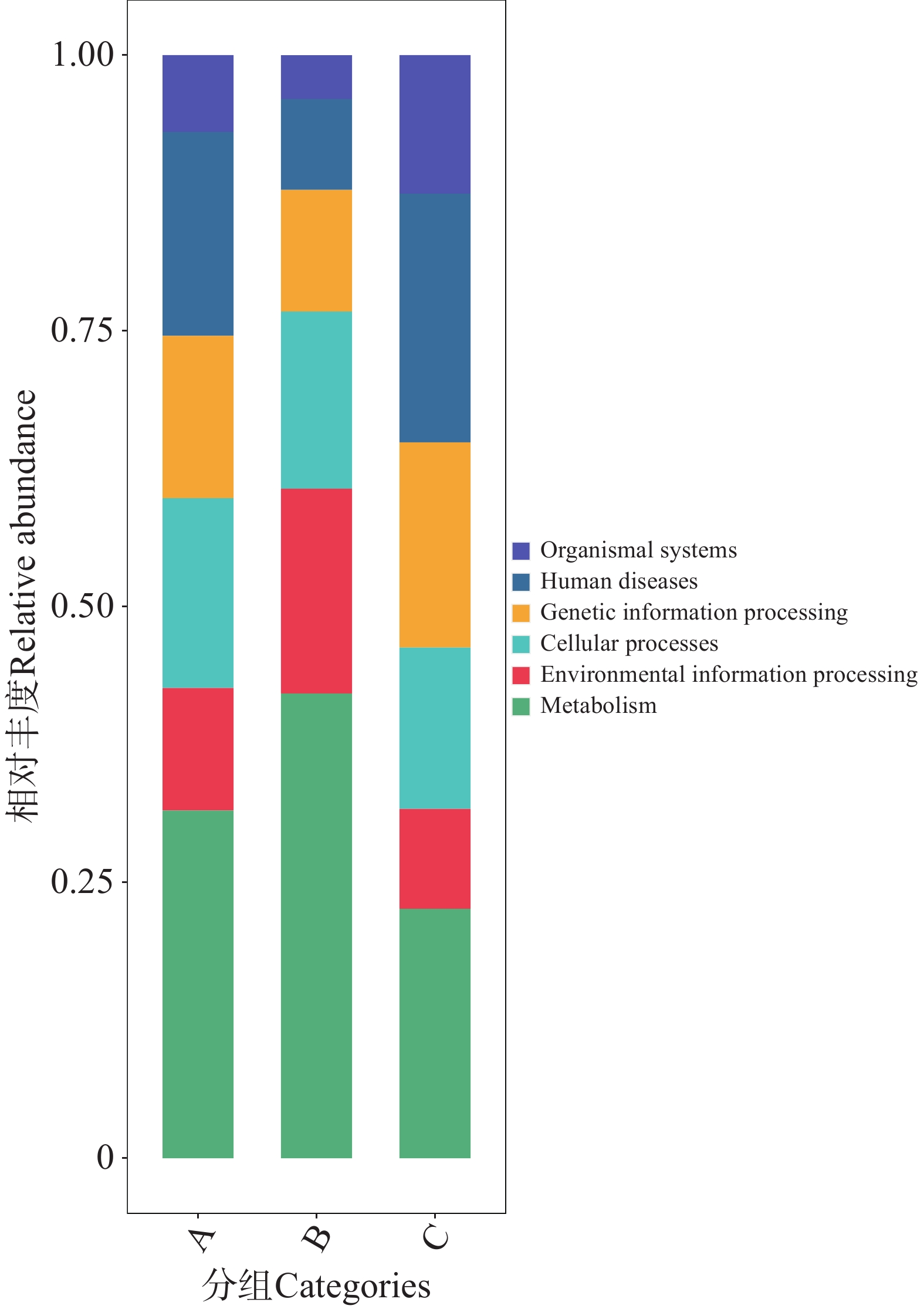
1)KEGG功能大类(Level A)的组间分布特征
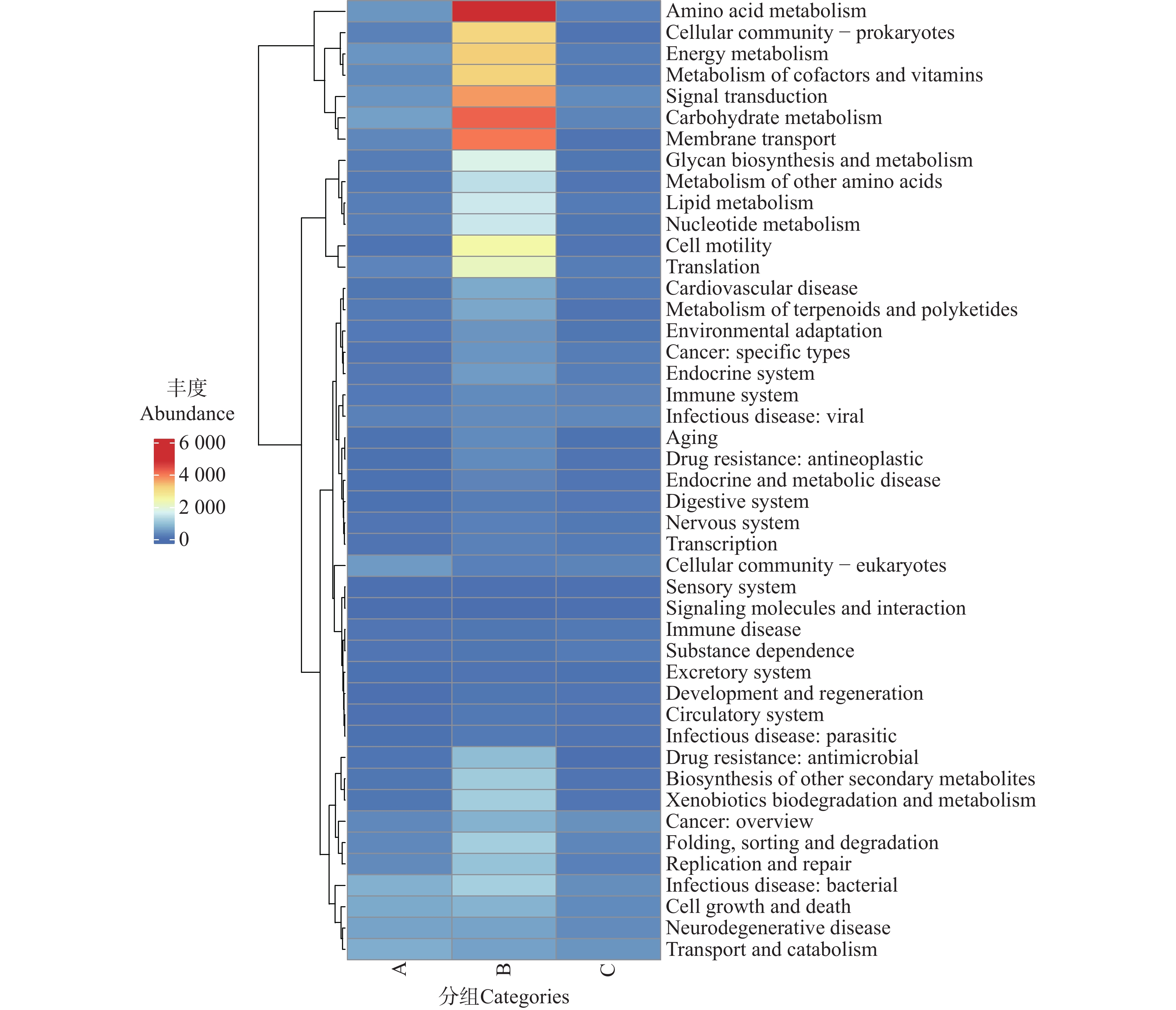
利用KEGG数据库对2 343 819个非冗余基因进行功能注释,共获得6个一级代谢通路,分别为代谢(Metabolism)、遗传信息处理(Genetic Information Processing)、环境信息处理(Environmental Information Processing)、细胞过程(Cellular Processes)、有机系统(Organismal Systems)和人类疾病(Human Diseases)通路(图8)。其中,基因功能相对丰度图显示(图9),B组代谢通路相较于A、C组的相对丰度更高,其中代谢通路在B组中相对丰度最高;而C组的人类疾病通路相对丰度更高。
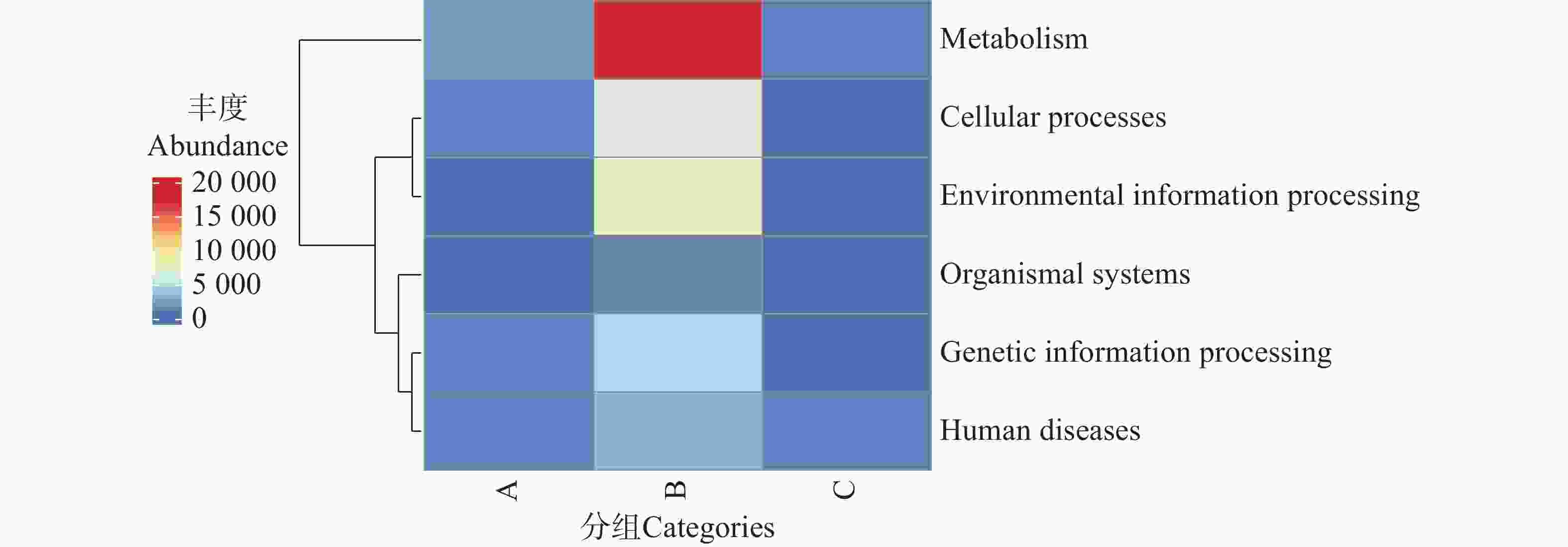
图 8 不同地区的蜱虫基因功能大类(Level A)热图
Figure 8. Heatmap of gene functional categories (Level A)of ticks from different regions
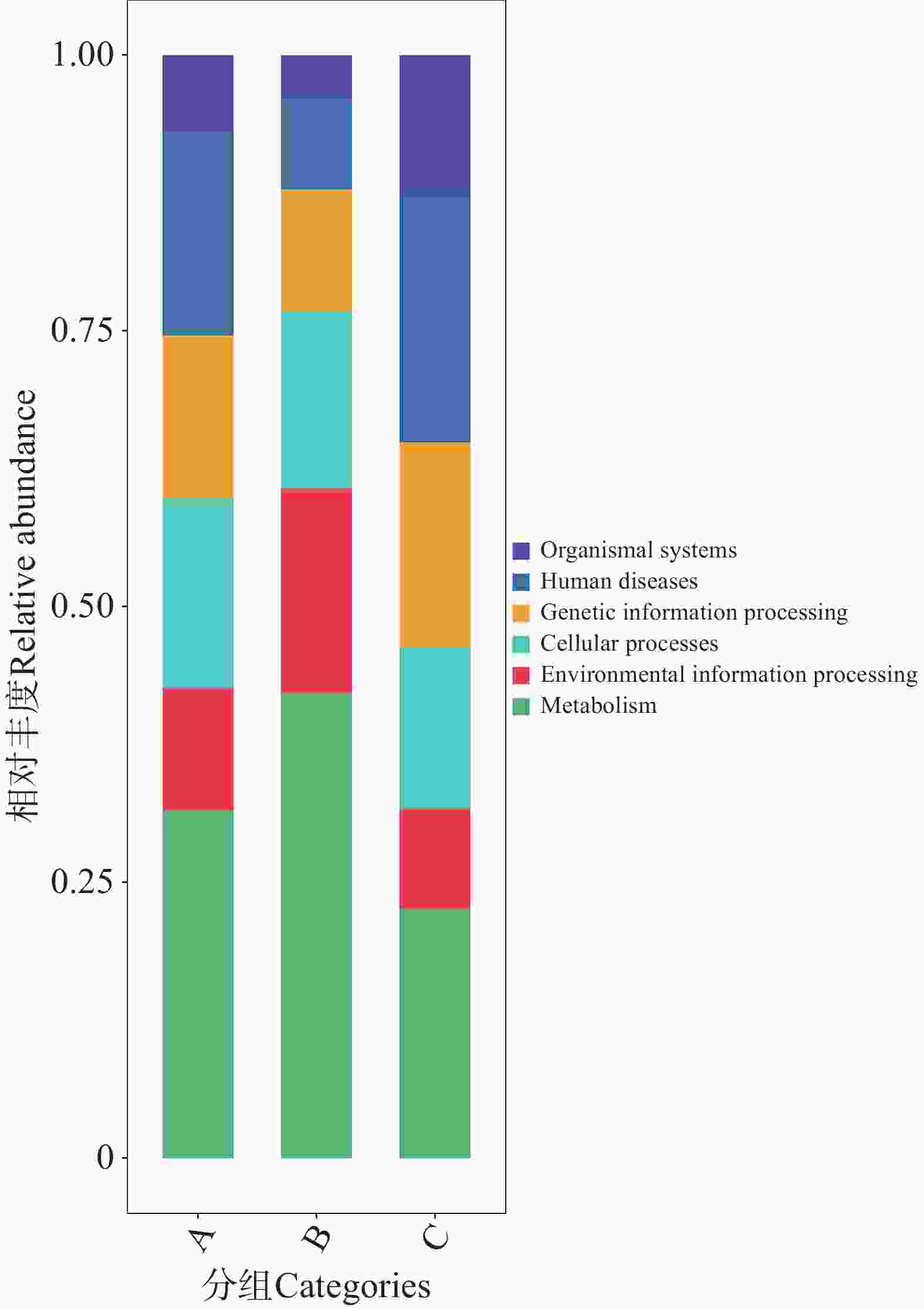
图 9 不同地区的蜱虫基因注释功能大类(Level A)相对丰度图
Figure 9. Relative abundance plot of gene-annotated functional categories (Level A)of ticks from different regions
2)KEGG功能中类(Level B)和具体通路(Level C)的组间分布特征
对KEGG Level B和KEGG Level C的相对丰度进行聚类热图和相对丰度图分析(图10~13)。结果表明,A、B、C三组在代谢通路、细胞群落互作、疾病相关通路等维度呈现显著差异。
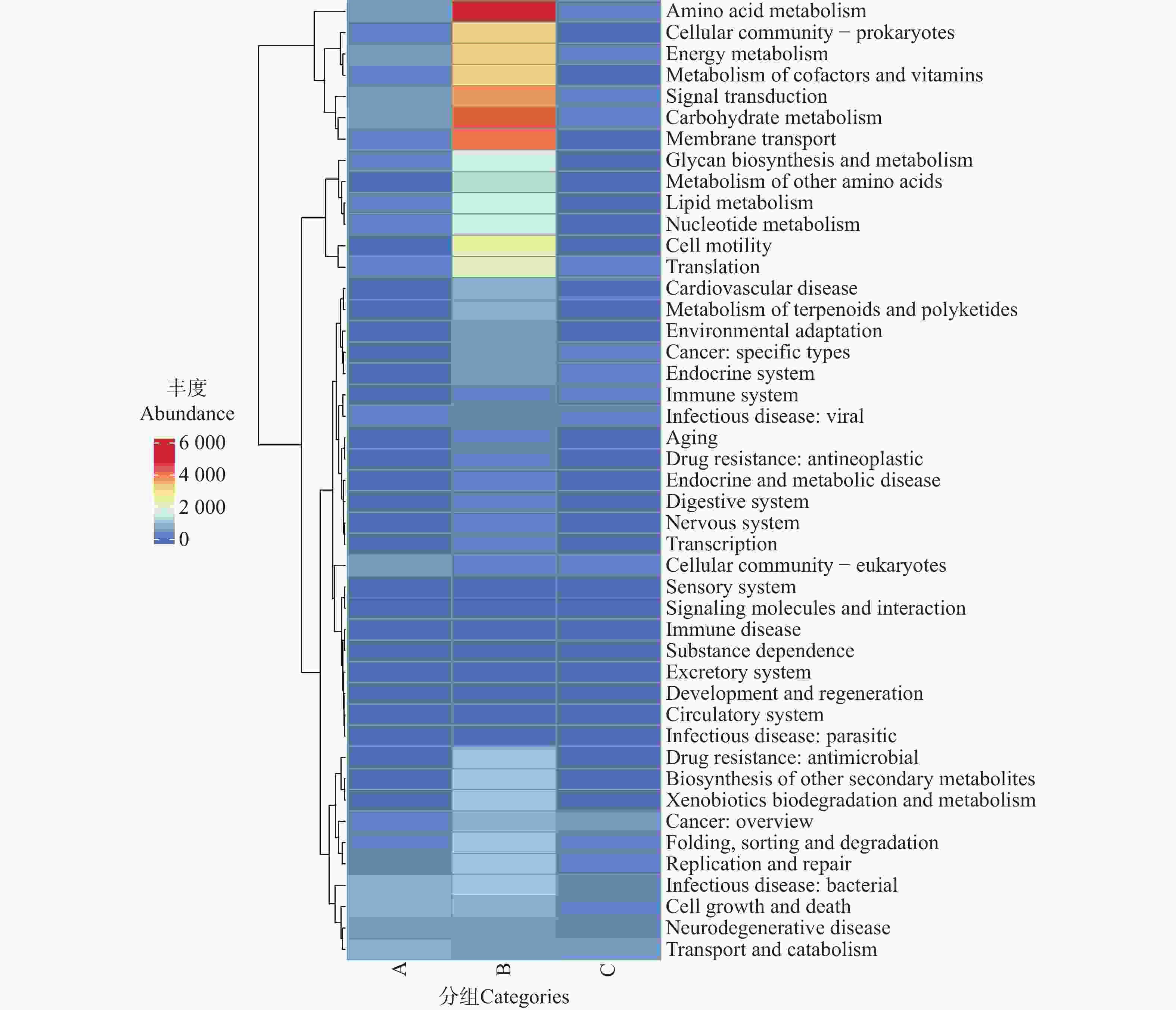
图 10 不同地区蜱虫基因注释功能中类(Level B)热图
Figure 10. Heatmap of gene-annotated functional subcategories (Level B)of ticks from different regions
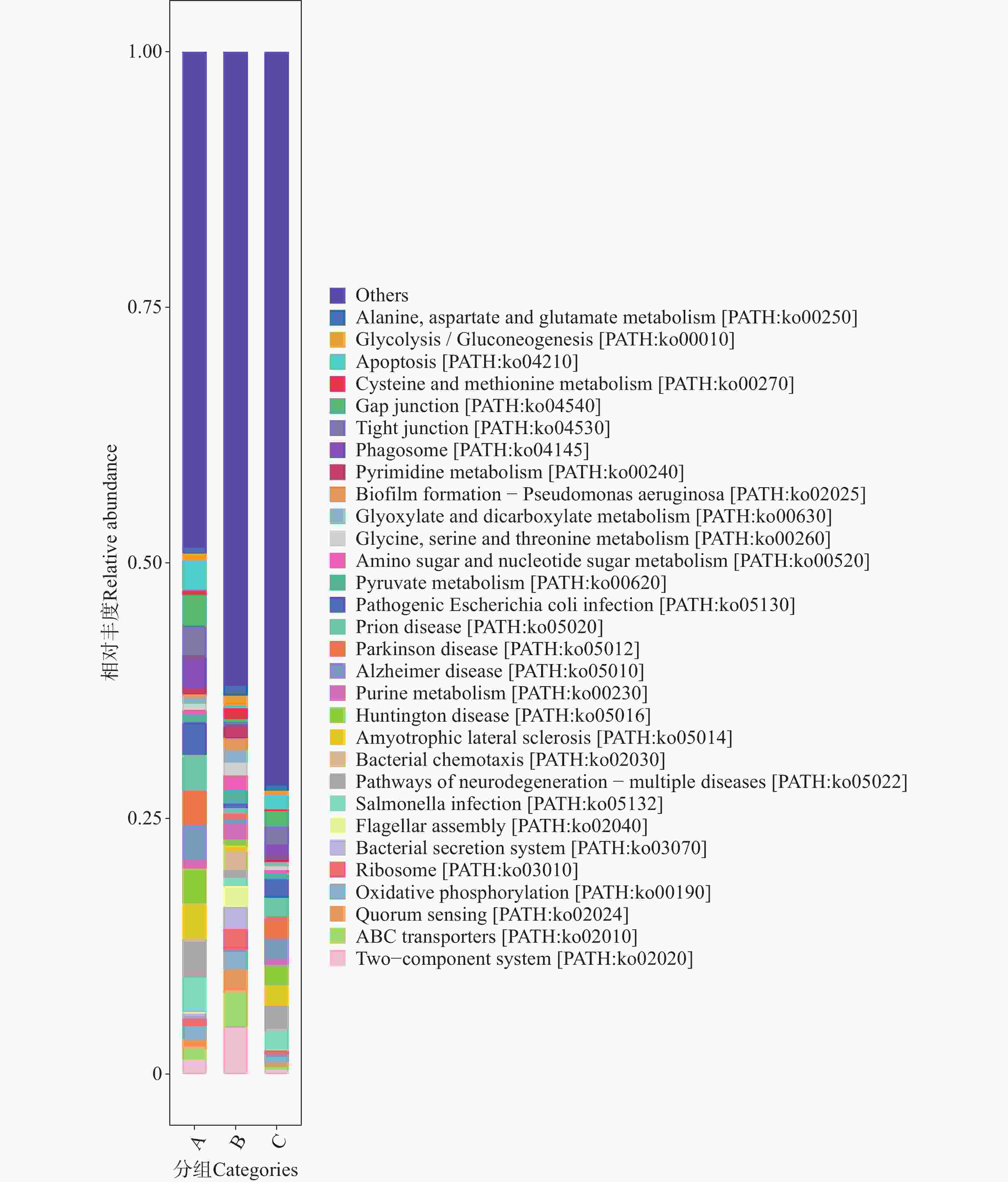
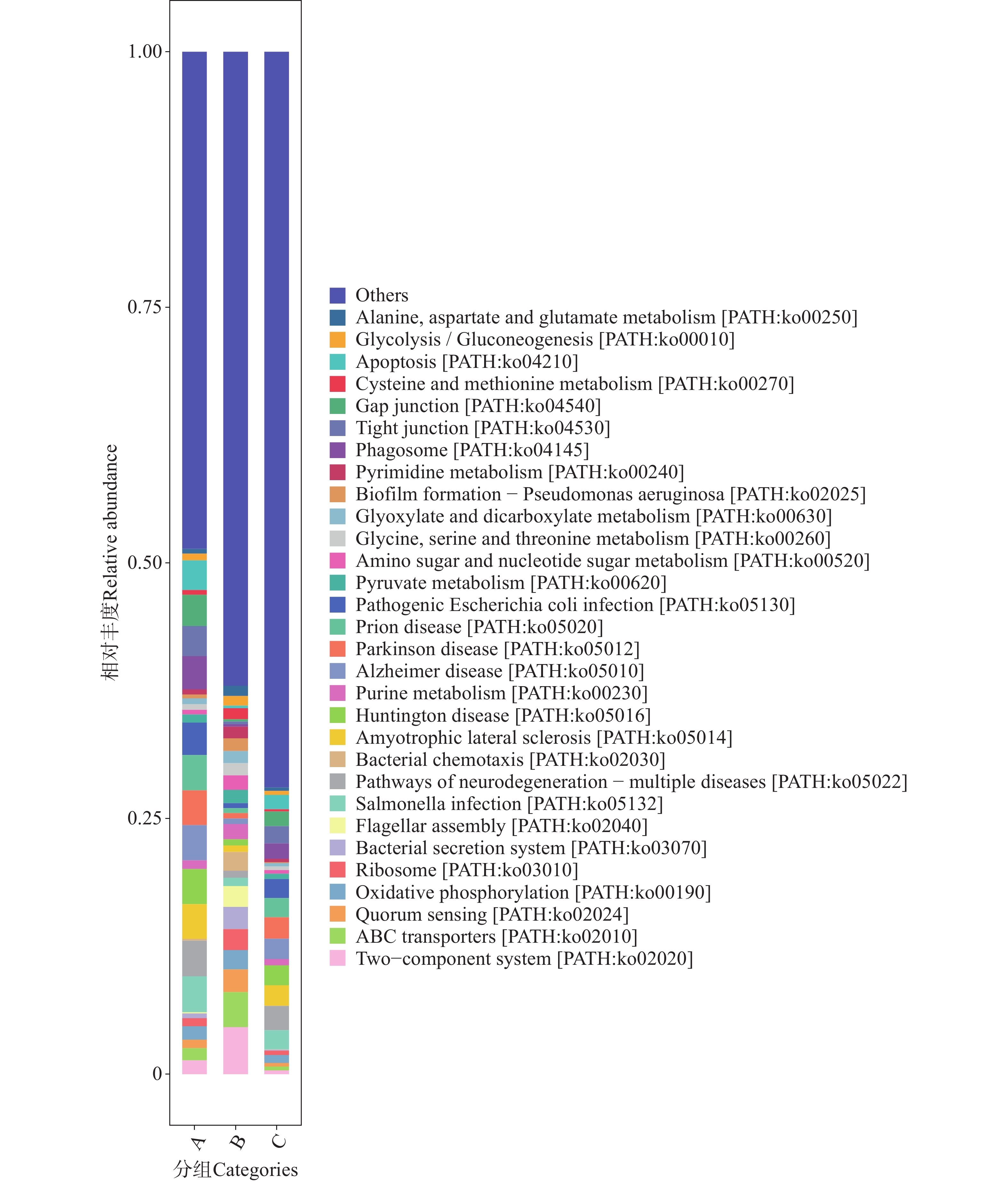
图 13 不同地区蜱虫基因注释功能具体通路(Level C)相对丰度图
Figure 13. Relative abundance plot of specific gene-annotated functional pathways (Level C)of ticks from different regions
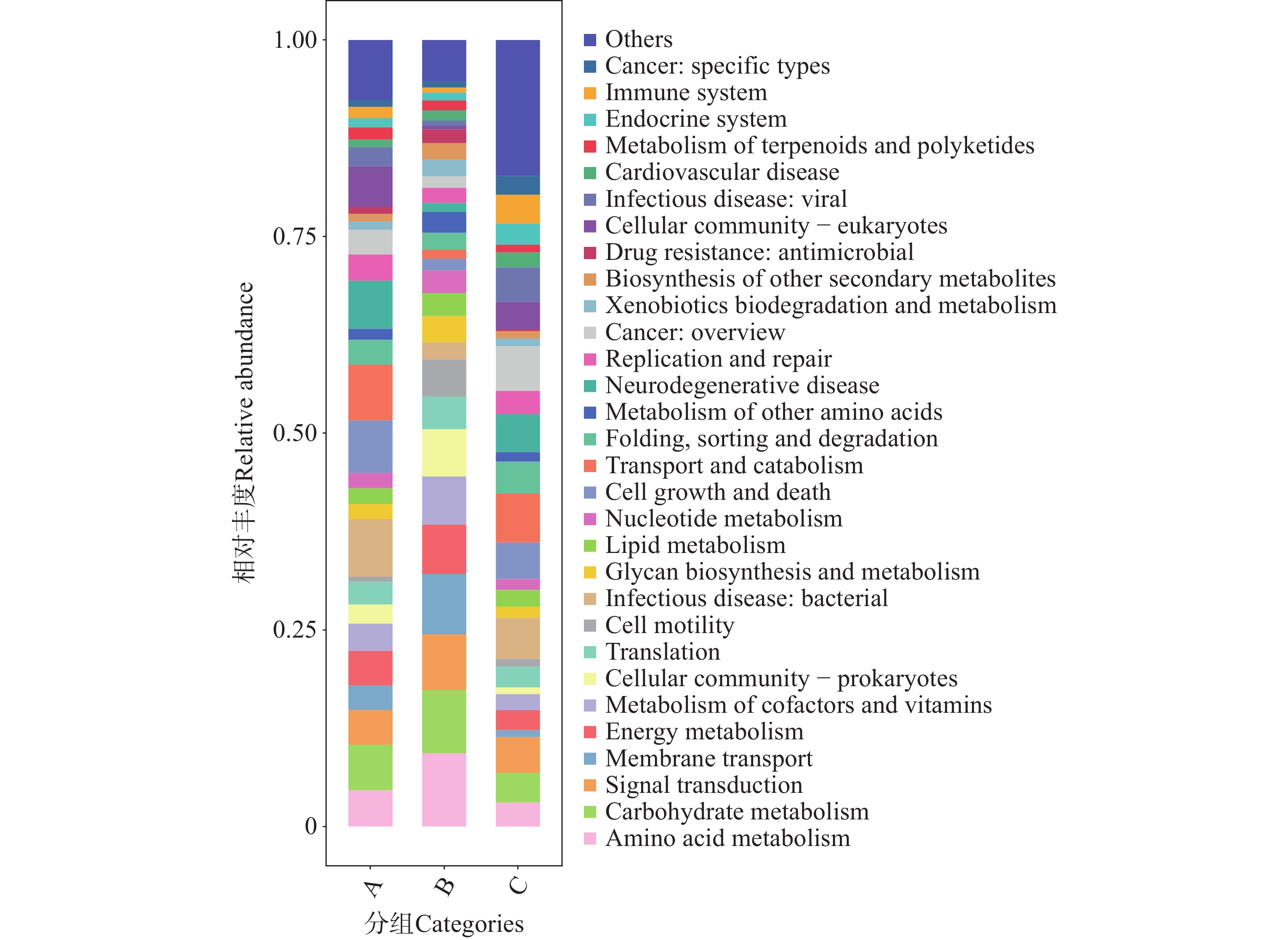
代谢通路方面,B组的代谢功能丰度显著高于A与C组,提示B微生物群落在能量转化与物质循环相关的核心代谢过程中具有更强的功能倾向性,反映该组微生物可能受特定选择压力驱动,向代谢功能富集的方向演替。B组同样在细胞过程与环境信息处理功能的丰度同样呈现相对优势。
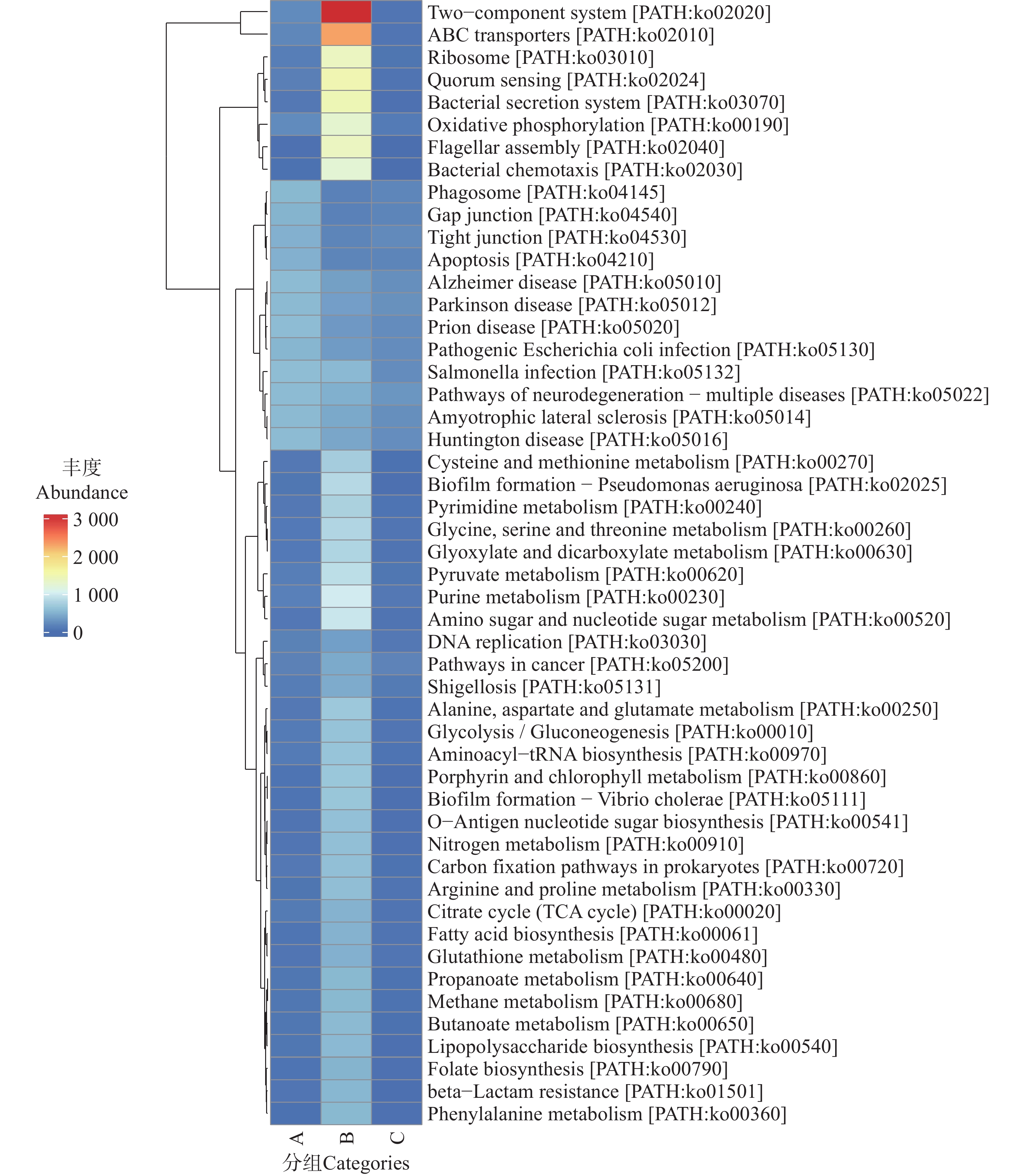
在人类疾病通路中,C组在病毒感染性疾病(Infectious disease viral)方面表现出较高的通路丰度。结合Level A功能富集的分析中,人类疾病相关功能的特征提示C组微生物群落可能携带更多与病毒感染相关的功能基因,存在潜在的病毒传播风险。在Level C的聚类热图(图12)与相对丰度柱状图(图13)中可见,B组在广谱抗菌药物耐药性及相关通路(如ABC transporters、Two-component system 等)中的丰度显著高于A组和C组。这些通路涉及 β-内酰胺类、大环内酯类、喹诺酮类等多种抗生素的耐药机制。例如,A、B、C三组的转运蛋白通路可通过多药外排泵基因主动将β-内酰胺类和大环内酯类抗生素排出细胞,减少药物在胞内积累[30];双组分系统通路则能够通过信号调控(如 PhoP/PhoQ 类系统)激活多药外排泵或改变膜通透性等相关基因的表达,从而降低胞内药物浓度[31]。此外,喹诺酮类药物的经典耐药机制还包括介导gyrA等耐药基因突变,降低药物与DNA旋转酶的结合效率以逃逸杀伤[32]。在抗生素生物合成方面,B组在抗生素生物合成及相关通路中的丰度显著高于A组和C组。这些通路涉及链霉素、四环素等抗生素的生物合成基因簇(如链霉素合成相关的 str 基因簇)。就细菌性感染疾病(Infectiousdisease bacterial)而言,在Level C代谢通路的相对丰度(图13)分析中,C组微生物群落中细菌性病原感染相关代谢通路更为富集,显著高于A组和B组。结合蜱虫常携带产气荚膜梭菌、破伤风梭菌等土传病原体,以及沙门氏菌、布鲁氏菌等动物源性病原体的特点,这些通路可能为细菌定植与侵袭宿主细胞提供代谢支持,使蜱虫更易成为细菌性病原的传播媒介,从而增加如沙门氏菌病、致病性大肠杆菌肠道感染等人兽共患细菌性疾病的流行风险。
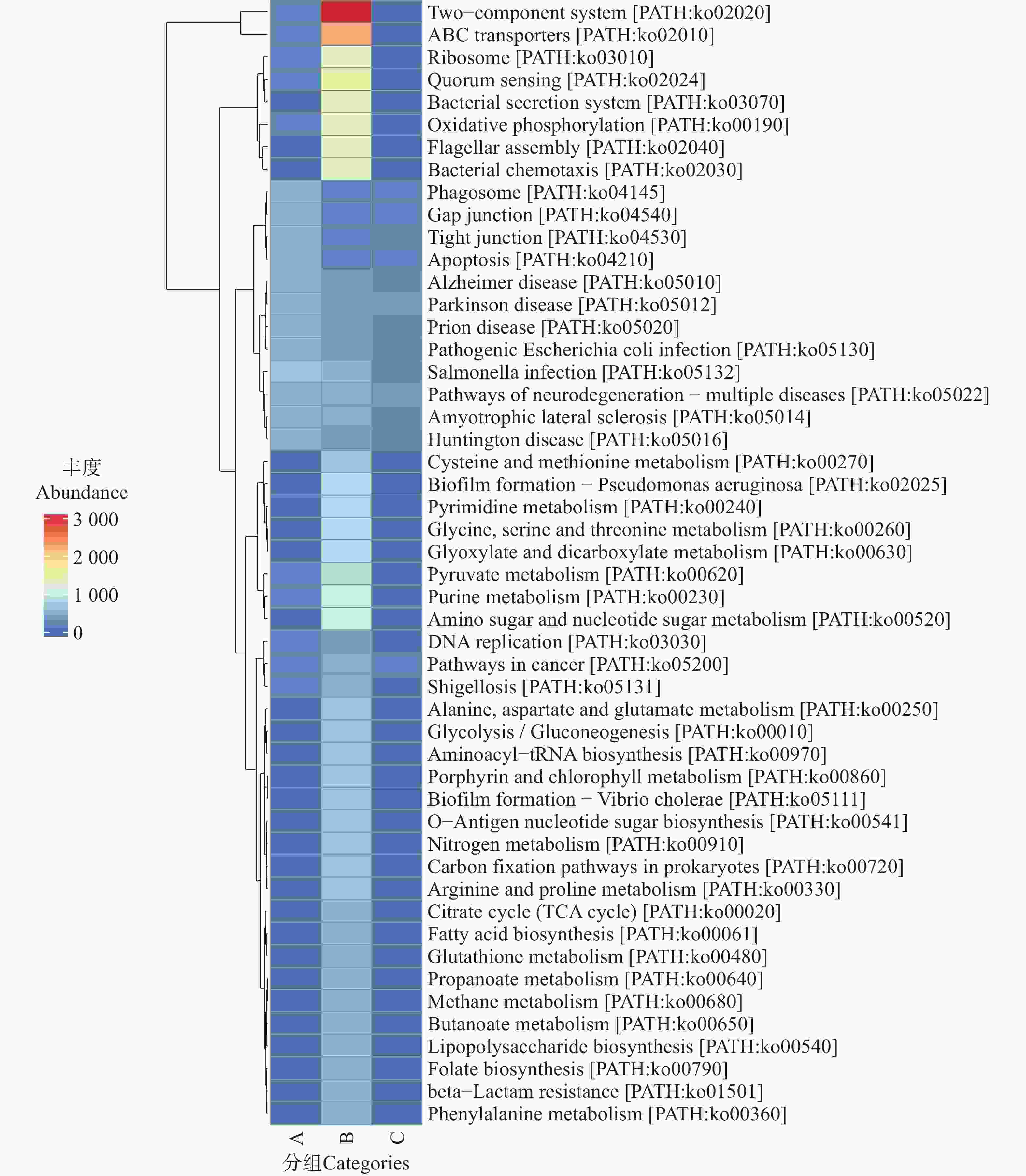
图 12 不同地区蜱虫基因注释具体通路(Level C)热图
Figure 12. Heatmap of specific gene-annotated functional pathways (Level C)of ticks from different regions
在寄生虫感染疾病方面,尽管整体通路丰度在热图中处于较低水平,C仍表现出相对较高的丰度值。
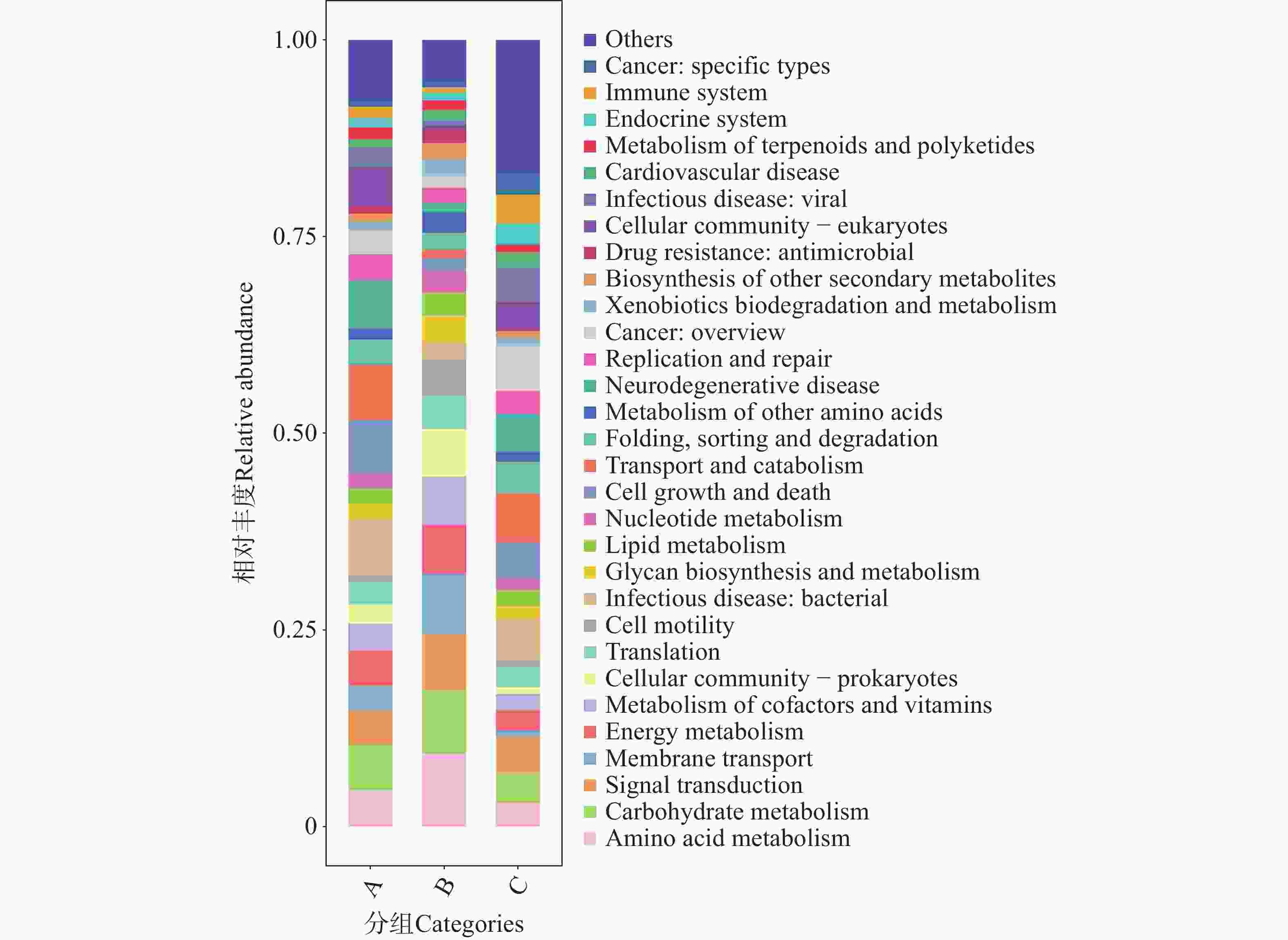
图 11 不同地区蜱虫基因注释功能中类相对丰度图
Figure 11. Relative abundance plot of gene-annotated functional subcategories (Level B)of ticks from different regions
-
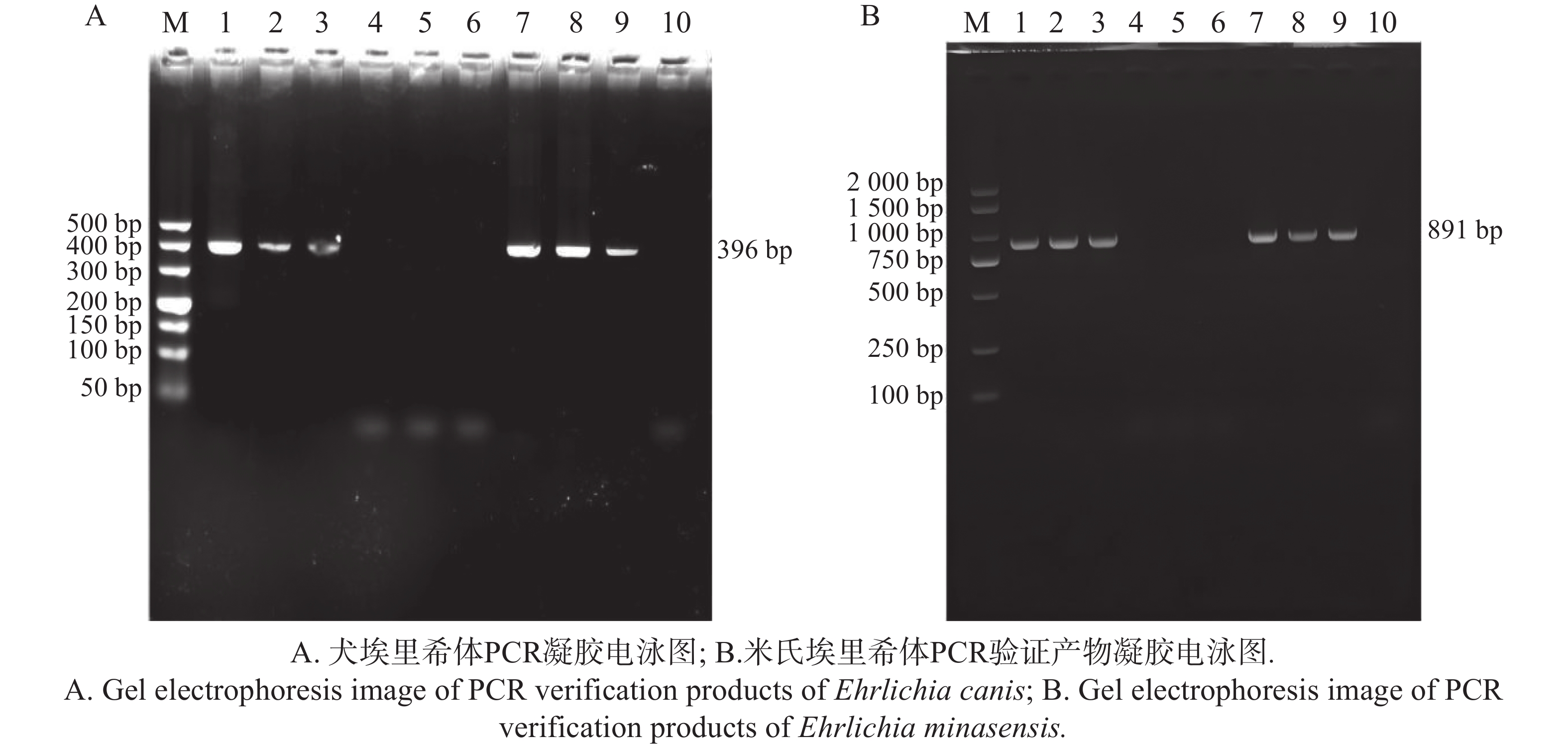
以埃里希体属 16S rRNA 基因与米氏埃里希体的 TRP36 基因为靶标进行 PCR 扩增后,将产物经 2%琼脂糖凝胶电泳分离,通过凝胶成像系统获取图像(图14)。结果显示,在3组9个样本中,查菲埃里希体(E. chaffeensis)与埃文埃里希体(E. ewingii)的检测结果均为阴性;(图14)中, 16S rRNA 基因扩增片段与标准分子量Marker相比,位于接近 400 bp 处;TRP36 基因扩增片段与标准分子量Marker相比,位于750~
1000 bp,两种基因的扩增产物条带大小均分别与预期大小396、891bp相符。将扩增产物测序后,将序列在 NCBI 数据库中进行比对,进一步确定为犬埃里希体和米氏埃里希体。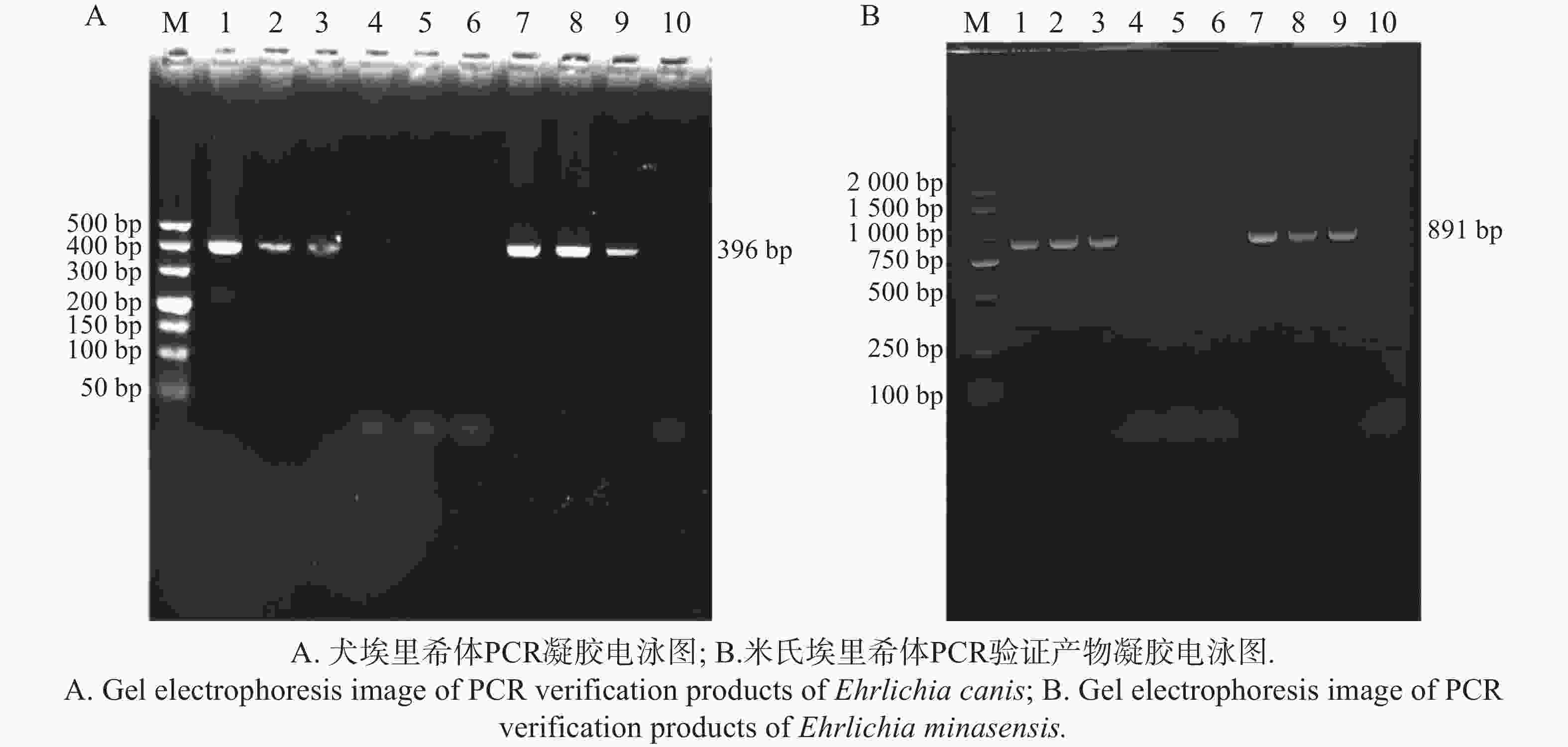
图 14 不同地区的蜱虫埃里希体属PCR鉴定产物凝胶电泳图
Figure 14. Gel electrophoresis images of PCR identification products of Ehrlichia spp. carried by ticks from different regions
-
蜱虫及其共生微生物的研究对于解析生态系统、发展病媒生物防控策略及研发新型诊断方法具有重要意义。然而,由于蜱虫及其体内微生物种类繁多、数量庞大,且部分类群难以通过传统培养方法进行研究,本研究采用宏基因组学技术,对来自海南省3个地区的蜱虫及其携带微生物进行了初步解析。本研究通过形态学结合 PCR 分子鉴定,明确定安县犬体表采集到的蜱虫为长角血蜱,白沙县猪体表、万宁市犬体表采集到的蜱虫为血红扇头蜱。从微生物群落结构的差异性特征来看,呈现组间差异显著高于组内;门水平上,细菌以变形菌门为绝对优势类群(相对丰度近 90%),真菌呈现蜱种关联差异(定安子囊菌门 50.79%,白沙、万宁毛霉门约 38%),病毒则表现出地理关联差异。种水平上,细菌共检出 533 种,以人兽共患病原为核心优势类群;真菌注释到 205 种,呈现蜱种与宿主环境关联差异;病毒共鉴定出 30 种,表现出地理与蜱种协同关联差异。在本研究中,埃里希体属中检测到犬埃里希体和米氏埃里希体,未检出查菲埃里希体等其他种类,与宏基因注释文件趋势一致;其中,米氏埃里希体在细菌整体里的相对丰度约30%,显著高于犬埃里希体。从基因功能分异结果来看,KEGG 功能分析显示 B 组代谢通路与耐药通路丰度显著高于 A、C 组,C 组人类疾病通路(病毒、细菌感染相关)更突出,反映不同环境中蜱虫微生物通过功能适配实现生存适应。综合分析显示,不同地区、不同宿主体表采集的不同蜱种,其体内微生物组成具有明显差异。细菌群落表现出与采集地区及宿主的强烈相关性,这与曹务春教授团队2025年发表的研究结论不同地区、蜱种携带的微生物存在显著差异相一致[7]。在三组样本中,A组、C组不仅细菌种类分布相似,其宿主动物类型也相同;而B组尽管蜱虫种类与C组相同,但宿主及其所处的生态环境均与A组、C组存在差异,最终导致其细菌群落的组成及分布与A组、C组表现出明显不同。
蜱虫微生物组的构成是一个动态过程,受到外部环境和吸血宿主的双重筛选。地理格局和气候条件通过影响植被、土壤微生物以及蜱虫自身的生理状态,对微生物组施加了基础性的选择压力[36]。宿主动物的血液为蜱虫提供了最直接的微生物来源,不同宿主体内独特的血液微生物会显著改变蜱虫摄食后的菌群[37]。综合研究表明,宿主种类和地理区域均是驱动其微生物组变异的独立且显著的因素。综上所述,本研究通过宏基因组学方法初步揭示了海南省不同地区蜱虫体内微生物群落的差异及其与蜱种、宿主的关联,首次在海南蜱虫中发现米氏埃里希体,明确了包括犬埃里希体和米氏埃里希体在内的埃里希体属在海南省犬源蜱虫中的分布。这些结果为蜱虫病、埃里希体病的精准防控、诊断靶标开发及相关生态研究提供了理论依据。但本研究的样本量及采样地点相对有限,主要集中于海南省3个区域,因此在空间覆盖性和生态代表性方面存在一定局限。未来研究可扩大采样范围、增加样本数量,并纳入与宿主相关的指标,以更全面、系统地揭示蜱虫微生物的组成及其功能与流行病学意义。
Metagenomic analysis of the microbial composition of ticks and the Genus Ehrlichia in the ticks in Hainan
-
摘要: 为了解海南省不同生境和不同动物体表蜱虫种类和蜱虫微生物组成特征及蜱传埃里希体属病原的种类和分布,本研究于海南省中部定安县(定安)、西部白沙黎族苗族自治县(白沙)和南部万宁市(万宁)3个市县采集猪和犬体表蜱虫,并经形态学和 PCR 鉴定蜱种,其中,在万宁犬体表采集的为长角血蜱;在定安犬体表和白沙猪体表采集到的为血红扇头蜱。结合宏基因组学分析蜱虫微生物组成、功能通路及蜱传埃里希体属种类,结果表明:1)在门水平上,3个市县的蜱虫体内细菌以变形菌门为主;蜱虫体内真菌中定安样本以子囊菌门为主,白沙与万宁样本以毛霉门为主;蜱虫体内病毒中定安样本以科索病毒门为优势种、白沙与万宁样本以负链 RNA 病毒门为优势种;2)在种水平上,检出伯氏柯克斯体、嗜吞噬细胞无形体和埃里希体等多种人兽共患病原,首次在海南蜱虫中检测到米氏埃里希体;3)功能通路呈组间分异,其中,白沙样本的蜱虫代谢及耐药通路丰度高,定安样本的蜱虫人类疾病通路突出。综合分析显示采集地点、蜱种、宿主对微生物群落影响显著。经PCR检测进一步验证,海南蜱虫携带的埃里希体属主要种类为犬埃里希体和米氏埃里希体。本研究结果为海南蜱虫病防控提供理论依据,同时为后续开发蜱传埃里希病的诊断方法提供数据支持。Abstract: To clarify the species of ticks on the body surface of animals, characteristics of tick microbial composition, as well as the species and distribution of tick-borne Ehrlichia pathogens in different habitats of Hainan Province, ticks were collected from the body surface of swine and dogs in three administrative regions of Hainan, namely Ding'an County (central Hainan), Baisha Li and Miao Autonomous County (western Hainan), and Wanning City (southern Hainan). Tick species were identified through morphological observation and PCR assay: Haemaphysalis longicornis was collected from dogs in Wanning, while Rhipicephalus sanguineus was isolated from dogs in Ding'an and swine in Baisha. Metagenomic approaches were further employed to analyze the microbial composition, functional pathways and species of tick-borne Ehrlichia in the collected ticks. The results showed that, Proteobacteria was the absolutely dominant bacterial phylum in ticks across the three regions. For fungi, Ascomycota was the dominant phylum in Ding'an ticks, whereas Mucoromycota predominated in ticks from Baisha and Wanning. In terms of viruses, Chrysoviridae was the dominant viral phylum in Ding'an ticks, while Negarnaviricota was the major one in Baisha and Wanning ticks. At the species level, a variety of important zoonotic pathogens were detected, including Coxiella burnetii, Anaplasma phagocytophilum and Ehrlichia spp., with Ehrlichia minasensis being identified in ticks from Hainan for the first time. The functional pathways of the tick microbial communities exhibited obvious intergroup divergence: ticks from Baisha showed high abundances of pathways related to metabolism and drug resistance, while those from Ding'an were characterized by a prominent enrichment of pathways associated with human diseases. Comprehensive analysis revealed that sampling location, tick species and host species exerted significant effects on the tick microbial community structure. Further verification via PCR assays confirmed that the dominant species of Ehrlichia carried by ticks in Hainan were E.canis and E. minasensis. The findings provide a theoretical basis for the prevention and control of tick-borne diseases in Hainan Province, and also offer fundamental data support for the subsequent development of diagnostic methods for tick-borne ehrlichiosis.
-
Key words:
- ticks /
- metagenomics /
- Ehrlichia /
- microbiome
-
图 2 3组蜱虫PCR鉴定产物凝胶电泳图
Fig. 2 Gel electrophoresis images of PCR identification products from three groups of ticks
图 3 不同地区的蜱虫门水平(A)和种水平(B)物种丰度数相似性分析图
Fig. 3 Species richness similarity analysis plots of ticks from different regions at the phylum level (Panel A)and species level (Panel B)
图 4 不同地区不同蜱虫门水平(A)和种水平(B)的主坐标分析
Fig. 4 Principal coordinate analysis (PCoA)plots of ticks from different regions at the phylum level (Panel A)and species level (Panel B)
图 5 不同地区的蜱虫携带的细菌门水平(A)和种水平(B)上的相对丰度
Fig. 5 Relative abundance of bacteria carried by ticks from different regions at the phylum level (Panel A)and species level (Panel B)
图 6 不同地区的蜱虫携带的真菌门水平(A)和种水平(B)上的相对丰度
Fig. 6 Relative abundance of fungi carried by ticks from different regions at the phylum level (Panel A)and species level
图 7 不同地区的蜱虫携带的病毒门水平(A)和种水平(B)上的相对丰度
Fig. 7 Relative abundance of viruses carried by ticks from different regions at the phylum level (Panel A)and species level
图 8 不同地区的蜱虫基因功能大类(Level A)热图
Fig. 8 Heatmap of gene functional categories (Level A)of ticks from different regions
图 9 不同地区的蜱虫基因注释功能大类(Level A)相对丰度图
Fig. 9 Relative abundance plot of gene-annotated functional categories (Level A)of ticks from different regions
图 10 不同地区蜱虫基因注释功能中类(Level B)热图
Fig. 10 Heatmap of gene-annotated functional subcategories (Level B)of ticks from different regions
图 13 不同地区蜱虫基因注释功能具体通路(Level C)相对丰度图
Fig. 13 Relative abundance plot of specific gene-annotated functional pathways (Level C)of ticks from different regions
图 12 不同地区蜱虫基因注释具体通路(Level C)热图
Fig. 12 Heatmap of specific gene-annotated functional pathways (Level C)of ticks from different regions
图 11 不同地区蜱虫基因注释功能中类相对丰度图
Fig. 11 Relative abundance plot of gene-annotated functional subcategories (Level B)of ticks from different regions
图 14 不同地区的蜱虫埃里希体属PCR鉴定产物凝胶电泳图
Fig. 14 Gel electrophoresis images of PCR identification products of Ehrlichia spp. carried by ticks from different regions
表 1 蜱虫样品信息及分组情况
Table 1 Sample Information and Grouping Details of Ticks
样本分组
Sample group样本名称
Sample name样本来源
Sample source采集宿主
Collection host采集状态
Collection status采集时间
Collection timeA A1 定安县新竹镇
Xinzhu Town, Ding'an犬
Canis未吸血,雌蜱
unfed female ticks2024-09-24 A2 A3 B B1 白沙黎族自治县打安镇可程村五指猪科技小院
Wuzhishan Breed Science and Technology
Courtyard, Kecheng Village, Da'an Town,
Baisha Li Autonomous County猪
Sus未吸血,雌蜱
unfed female ticks2025-01-11 B2 B3 C C1 万宁市兴隆镇华侨农场
Xinglong Overseas Chinese Farm,
Xinglong Town, Wanning City犬
Canis未吸血,雌蜱
unfed female ticks2025-04-09 C2 C3  下载: 导出CSV
下载: 导出CSV
表 2 血红扇头蜱和长角血蜱PCR引物序列
Table 2 PCR primer Sequences of Rhipicephalus sanguineus and Haemaphysalis longicornis
引物名称
Primer name引物序列(5'—3')
Primer sequence(5'—3')扩增长度/bp
Amplicon length /bp物种
Species参考文献
ReferencesRs-16S-F CTGCTCAATGATTTTTTAAATTGCTGTGG 460 Rhipicephalus sanguineus [11] Rs-16S-R CCGGTCTGAACTCAGATCAAGT 460 Rhipicephalus sanguineus [11] Nad-5-F TCTAAAATTAAATCCTTTGAAT 500 Haemaphysalis longicornis [12] Nad-5-R AAGAGCCCAAATTCCATTTTC
下载: 导出CSV
表 3 埃里希体属PCR引物序列
Table 3 PCR primer Sequences of the Ehrlichia Genus
引物名称
Primer name引物序列(5—3')
Primer sequence(5'—3')扩增长度 /bp
Amplicon length /bp物种
Species参考文献
ReferencesTRP36-F2 TTTAAAACAAAATTAACACACTA 891 E. minasensis [13] TRP36-R1 AAGATTAACTTAATACTCAATATTACT ECC AGAACGAACGCTGGCGGCAAGC 477 Ehrlichia Spp 16s [14] ECB CGTATTACCGCGGCTGCTGGCA ECAN5 CAATTATTTATAGCCTCTGGCTATAGGA 396 E. canis HE3 TATAGGTACCGTCATTATCTTCCCTAT HE1 CAATTGCTTATAACCTTTTGGTTATAAAT 396 E .chaffeensis HE3 TATAGGTACCGTCATTATCTTCCCTAT EE52 CGAACAATTCCTAAATAGTCTCTGAC 396 E. ewengii HE3 TATAGGTACCGTCATTATCTTCCCTAT
下载: 导出CSV
表 4 海南3个不同地区采集蜱虫宏基因测序数据
Table 4 Metagenomic Sequencing Data of Ticks Collected from Three Different Regions in Hainan
样本分组
Sample group样本名称
Sample game原始数据(G)
Raw data(G)有效数据(G)
Valid data(G)有效数据率(%)
Valid data rate(%)GC(%)
GC content(%)Q30% A A1 9.87 9.01 91.17 47.65 96.39 A2 9.42 8.62 A3 10.60 9.62 B B1 10.27 9.37 90.31 50.12 95.97 B2 8.83 7.95 B3 9.48 8.50 C C1 14.94 10.67 71.18 47.08 96.51 C2 8.89 6.53 C3 10.59 7.30 合计92.89 合计77.57 注:GC占比为有效数据中两种碱基占总碱基的占比;Q30是测序数据中有效数据质量值大于或等于30的碱基所占的百分比。 Note:GC content refers to the proportion of guanine and cytosine bases relative to the total bases in the valid data; Q30 represents the percentage of bases in the valid sequencing data with a quality value greater than or equal to 30.
下载: 导出CSV
-
[1] Philippe C, Denis L A, Fonville M, et al. Diversity of the Ixodes ricinus Microbiome across Belgian Ecoregions and its association with pathogen and Symbiont presence [J]. Microbial Ecology, 2025, 88(1): 73. https://doi.org/10.1007/s00248-025-02571-8 doi: 10.1007/s00248-025-02571-8 [2] Sui L, Wang W F, Guo X R, et al. Multi-protomics analysis identified host cellular pathways perturbed by tick-borne encephalitis virus infection [J]. Nature Communications, 2024, 15(1): 10435. https://doi.org/10.1038/s41467-024-54628-w doi: 10.1038/s41467-024-54628-w [3] Zhang H, Wang Y N, Chen C G, et al. A novel rapid visual nucleic acid detection technique for tick-borne encephalitis virus by combining RT-recombinase-aided amplification and CRISPR/Cas13a coupled with a lateral flow dipstick [J]. International Journal of Biological Macromolecules, 2024, 275: 133720. https://doi.org/10.1016/j.ijbiomac.2024.133720 doi: 10.1016/j.ijbiomac.2024.133720 [4] de la Fuente J, Agustin Estrada-Pena A, Venzal J M, et al. Overview: ticks as vectors of pathogens that cause disease in humans and animals [J]. Frontiers in Bioscience, 2008, 13: 6938−6946. https://doi.org/10.2741/3200 doi: 10.2741/3200 [5] Fang L Q, Liu K, Li X L, et al. Emerging tick-borne infections in mainland China: an increasing public health threat [J]. The Lancet Infectious Diseases, 2015, 15(12): 1467−1479. https://doi.org/10.1016/S1473-3099(15)00177-2 doi: 10.1016/S1473-3099(15)00177-2 [6] Zhao G P, Wang Y X, Fan Z W, et al. Mapping ticks and tick-borne pathogens in China [J]. Nature Communications, 2021, 12(1): 1075. https://doi.org/10.1038/s41467-021-21375-1 doi: 10.1038/s41467-021-21375-1 [7] Du L F, Shi W Y, Cui X M, et al. Genome-resolved metagenomics reveals microbiome diversity across 48 tick species [J]. Nature Microbiology, 2025, 10(10): 2631−2645. https://doi.org/10.1038/s41564-025-02119-z doi: 10.1038/s41564-025-02119-z [8] Mitchell J K, Matthee S, Ndhlovu A, et al. The Microbiome and Coxiella diversity found in Amblyomma hebraeum and Dermacentor rhinocerinus ticks sampled from white rhinoceros [J]. Microbial Ecology, 2025, 88(1): 48. https://doi.org/10.1007/s00248-025-02549-6 doi: 10.1007/s00248-025-02549-6 [9] 陆宝麟, 吴永厚. 中国重要医学昆虫分类与鉴别[M]. 郑州: 河南科学技术出版社, 2003. (查阅网上资料, 未找到页码信息, 请确认补充) [10] 叶向光. 常见医学蜱螨图谱[M]. 北京: 科学出版社, 2020. (查阅网上资料, 未找到页码信息, 请确认补充) [11] 安丽萍, 裴宇霄, 索鹏辉, 等. 海南地区血红扇头蜱的生物学特性及对宿主的影响[J]. 中国热带医学, 2022, 22(6): 522−528. https://doi.org/10.13604/j.cnki.46-1064/r.2022.06.07 doi: 10.13604/j.cnki.46-1064/r.2022.06.07 [12] 李中波, 尹会方, 程天印, 等. 长角血蜱信阳株和浏阳株的ITS-1、nad5基因序列特征分析[J/OL]. 中国动物传染病学报, 1−8(2025-02-25)[2025-11-26]. https://doi.org/10.19958/j.cnki.cn31-2031/s.20250225.001 [13] Black W C, Piesman J. Phylogeny of hard- and soft-tick taxa (Acari: Ixodida) based on mitochondrial 16S rDNA sequences [J]. Proceedings of the National Academy of Sciences of the United States of America, 1994, 91(21): 10034−10038. https://doi.org/10.1073/pnas.91.21.10034 doi: 10.1073/pnas.91.21.10034 [14] Murphy G L, Ewing S A, Whitworth L C, et al. A molecular and serologic survey of Ehrlichia canis, E. chaffeensis, and E. ewingii in dogs and ticks from Oklahoma [J]. Veterinary Parasitology, 1998, 79(4): 325−339. https://doi.org/10.1016/S0304-4017(98)00179-4 doi: 10.1016/S0304-4017(98)00179-4 [15] Oba P M, Carroll M Q, Alexander C, et al. Microbiota populations in supragingival plaque, subgingival plaque, and saliva habitats of adult dogs [J]. Animal Microbiome, 2021, 3(1): 38. https://doi.org/10.1186/s42523-021-00100-9 doi: 10.1186/s42523-021-00100-9 [16] Swanson K S, Dowd S E, Suchodolski J S, et al. Phylogenetic and gene-centric metagenomics of the canine intestinal microbiome reveals similarities with humans and mice [J]. The ISME Journal, 2011, 5(4): 639−649. https://doi.org/10.1038/ismej.2010.162 doi: 10.1038/ismej.2010.162 [17] Thomas R J, Dumler J S, Carlyon J A. Current management of human granulocytic anaplasmosis, human monocytic ehrlichiosis and Ehrlichia ewingii ehrlichiosis [J]. Expert Review of Anti-infective Therapy, 2009, 7(6): 709−722. https://doi.org/10.1586/eri.09.44 doi: 10.1586/eri.09.44 [18] Ahaduzzaman M, Reza M M B. Global and regional seroprevalence of coxiellosis in small ruminants: a systematic review and meta‐analysis [J]. Veterinary Medicine and Science, 2024, 10(3): e1441. https://doi.org/10.1002/vms3.1441 doi: 10.1002/vms3.1441 [19] Stevens D L, Bisno A L, Chambers H F, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the infectious diseases society of America [J]. Clinical Infectious Diseases, 2014, 59(2): e10−e52. https://doi.org/10.1093/cid/ciu296 doi: 10.1093/cid/ciu296 [20] Kanamori H, Aoyagi T, Kuroda M, et al. Chromobacterium haemolyticum pneumonia associated with near-drowning and river water, Japan [J]. Emerging Infectious Diseases, 2020, 26(9): 2186−2189. https://doi.org/10.3201/eid2609.190670 doi: 10.3201/eid2609.190670 [21] Cabezas-Cruz A, Valdés J J, de la Fuente J. The glycoprotein TRP36 of Ehrlichia sp. UFMG-EV and related cattle pathogen Ehrlichia sp. UFMT-BV evolved from a highly variable clade of E. canis under adaptive diversifying selection [J]. Parasites & Vectors, 2014, 7: 584. https://doi.org/10.1186/s13071-014-0584-5 doi: 10.1186/s13071-014-0584-5 [22] Bouwknegt M, van Dorp S, Kuijper E. Burden of Clostridium difficile infection in the United States [J]. New England Journal of Medicine, 2015, 372(24): 2368−2370. https://doi.org/10.1056/NEJMc1505190 doi: 10.1056/NEJMc1505190 [23] Lister P D, Wolter D J, Hanson N D. Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms [J]. Clinical Microbiology Reviews, 2009, 22(4): 582−610. https://doi.org/10.1128/CMR.00040-09 doi: 10.1128/CMR.00040-09 [24] Pfohl-Leszkowicz A, Manderville R A. Ochratoxin A: an overview on toxicity and carcinogenicity in animals and humans [J]. Molecular Nutrition & Food Research, 2007, 51(1): 61−99. https://doi.org/10.1002/mnfr.200600137 doi: 10.1002/mnfr.200600137 [25] Kauffman C A. Histoplasmosis: a Clinical and Laboratory Update [J]. Clinical Microbiology Reviews, 2007, 20(1): 115−132. https://doi.org/10.1128/CMR.00027-06 doi: 10.1128/CMR.00027-06 [26] Mansfield K L, Jizhou L, Phipps L P, et al. Emerging tick-borne viruses in the twenty-first century [J]. Frontiers in Cellular and Infection Microbiology, 2017, 7: 298. https://doi.org/10.3389/fcimb.2017.00298 doi: 10.3389/fcimb.2017.00298 [27] Yu X J, Liang M F, Zhang S Y, et al. Fever with thrombocytopenia associated with a novel bunyavirus in China [J]. New England Journal of Medicine, 2011, 364(16): 1523−1532. https://doi.org/10.1056/NEJMoa1010095 doi: 10.1056/NEJMoa1010095 [28] Walker P J, Firth C, Widen S G, et al. Evolution of genome size and complexity in the Rhabdoviridae [J]. PLoS Pathogens, 2015, 11(2): e1004664. https://doi.org/10.1371/journal.ppat.1004664 doi: 10.1371/journal.ppat.1004664 [29] Jia N, Liu H B, Ni X B, et al. Emergence of human infection with Jingmen tick virus in China: a retrospective study [J]. EBioMedicine, 2019, 43: 317−324. https://doi.org/10.1016/j.ebiom.2019.04.004 doi: 10.1016/j.ebiom.2019.04.004 [30] Piddock L J V. Multidrug-resistance efflux pumps? Not just for resistance [J]. Nature Reviews Microbiology, 2006, 4(8): 629−636. https://doi.org/10.1038/nrmicro1464 doi: 10.1038/nrmicro1464 [31] Gunn J S. The Salmonella PmrAB regulon: lipopolysaccharide modifications, antimicrobial peptide resistance and more [J]. Trends in Microbiology, 2008, 16(6): 284−290. https://doi.org/10.1016/j.tim.2008.03.007 doi: 10.1016/j.tim.2008.03.007 [32] Hooper D C, Jacoby G A. Mechanisms of drug resistance: quinolone resistance [J]. Annals of the New York Academy of Sciences, 2015, 1354(1): 12−31. https://doi.org/10.1111/nyas.12830 doi: 10.1111/nyas.12830 [33] 唐菁. 蜱传立克次体扩增子测序方法的建立及其初步应用[D]. 昆明: 昆明理工大学, 2024. https://doi.org/10.27200/d.cnki.gkmlu.2024.000923 [34] Li J J, Liu X X, Mu J Q, et al. Emergence of a novel Ehrlichia minasensis strain, harboring the major immunogenic glycoprotein trp36 with unique tandem repeat and C-terminal region sequences, in Haemaphysalis hystricis ticks removed from free-ranging sheep in Hainan province, China [J]. Microorganisms, 2019, 7(9): 369. https://doi.org/10.3390/microorganisms7090369 doi: 10.3390/microorganisms7090369 [35] Moura De Aguiar D, Pessoa Araújo Junior J, Nakazato L, et al. Isolation and characterization of a novel pathogenic strain of Ehrlichia minasensis [J]. Microorganisms, 2019, 7(11): 528. https://doi.org/10.3390/microorganisms7110528 doi: 10.3390/microorganisms7110528 [36] Van Treuren W, Ponnusamy L, Brinkerhoff R J, et al. Variation in the Microbiota of Ixodes ticks with regard to geography, species, and sex [J]. Applied and Environmental Microbiology, 2015, 81(18): 6200−6209. https://doi.org/10.1128/AEM.01562-15 doi: 10.1128/AEM.01562-15 [37] Ross B D, Hayes B, Radey M C, et al. Ixodes scapularis does not harbor a stable midgut microbiome [J]. The ISME Journal, 2018, 12(11): 2596−2607. https://doi.org/10.1038/s41396-018-0161-6 doi: 10.1038/s41396-018-0161-6 -
 点击查看大图
点击查看大图
计量
- 文章访问数: 413
- HTML全文浏览量: 221
- 被引次数: 0
