

-
水稻(Oryza sativa)是全世界最重要的粮食作物之一,为全世界一半以上的人口提供食物,所以其产量和抗病性一直是研究的热点[1]。杂交水稻产量虽高,但是病虫害仍是影响产量进一步提高的主要因素之一,比如稻瘟病、白叶枯病[2]等,每年因病害造成的减产约为15%~30%。水稻类病斑(rice lesion mimic)突变体是指在无外源病原菌、无逆境无损伤的情况下,自发在水稻叶片或其他部位产生坏死斑表型的突变体,是植物超敏反应(hypersensitive response, HR)引起的一种程序性细胞死亡(programmed cell death, PCD),与由病原菌感染引起的病变非常相似[1, 3]。类病斑的形成机制颇为复杂,涉及多种生物学过程,如蛋白质的磷酸化与去磷酸化、泛素化修饰、活性氧(reactive oxygen species, ROS)的生成、脂肪酸代谢途径、丝裂原活化蛋白激酶(MAPK)信号级联反应及离子转运等。植物类病斑基因突变所导致的生理改变呈现多面性,包括生长发育、代谢活动及抗病机制等多个层面。在已报道的200多种水稻类病斑突变体中,水稻类病斑突变体的表型大多是单个隐性核基因发生突变造成的,如spl4[4]、spl21[5]和spl30[6]等,少数由显性基因控制,如Spl18[7],nls1-1D[8]、spl-D[9]、Spl26[10]和LIL1[11]。大多数水稻类病斑突变体都表现出对一个或多个病原菌具有抗病性,因此,水稻类病斑突变体成为水稻抗病分子机制研究和抗病育种的重要种质资源[12]。现代遗传学中,精准鉴定与特定性状相关的基因至关重要。传统图位克隆方法虽有效但耗时且成本高。随着二代测序技术(next-generation sequencing, NGS)发展,集群分离分析法(bulked segregant analysis, BSA)和突变位点图谱(MutMap)方法出现,提供了高效经济的基因定位新途径[13 − 14]。BSA和全基因组重测序相结合,通过选择目标性状的极端表型个体构建混合样本池并测序,快速鉴定与目标性状连锁的突变位点[15]。MutMap是BSA的具体应用,适用于甲基磺酸乙酯(ethyl methanesulfonate,EMS)诱变隐性突变基因分析,通过计算突变位点频率确定连锁染色体区段和突变位点[16 − 19]。
本研究团队从经EMS诱变后的籼稻‘黄华占’(‘HHZ’)群体中发现了1个类病斑突变体,命名为LMM43。以HHZ野生型为对照,来研究LMM43突变体的特性。选择‘HHZ’× LMM43杂交得到的F2群体为实验材料,利用BSA-seq对类病斑进行基因定位分析,旨在挖掘新型的抗病育种的重要基因,为进一步理解水稻抗病机制和培育抗病品种提供新的理论依据[20]。
-
本实验中所用到的水稻材料有籼稻(Oryza sativa ssp.)‘黄华占’(‘HHZ’),以及经过EMS诱变筛选出‘HHZ’背景的LMM43突变体,已经过多代自交获得了稳定遗传的表型。
-
将LMM43突变体及‘HHZ’野生型(wild type, WT)水稻种植于海南省三亚市崖州区独村实验中心基地(109º18′N, 18º36′E),进行常规水肥管理。观察在全生育期内LMM43和WT植株的表型差异,记录LMM43突变体类病斑的出现情况,关注其斑点出现的时间点、形状、颜色、位置、密度等。在拔节期(60 d)取叶片用于BSA-seq测序的极端性状样本池构建。
-
为确定该突变体的遗传模式,以‘HHZ’作为母本,LMM43作为父本进行杂交,该工作于陵水试验基地完成。收获的F1代种子,种植后观察F1代植株表型,将杂株排除后自交获得F2代种子。继续将F2代群体种植于独村实验中心基地获得遗传分离群体,对其进行观察和取样工作,统计性状分离情况。
-
选取了白叶枯病菌的7个生理小种:PXO61、PXO86、PXO79、PXO71、PXO112、PXO99和PXO145。使用剪叶法分别对野生型和突变体进行接菌处理[21],每个生理小种接种3株水稻,20 d后对接种叶片的病斑长度进行拍照及测量。
-
在F2代群体处于拔节期时,分别选择类病斑表型和野生型表型各30株及LMM43突变体亲本和HHZ亲本各2株的叶片,提取DNA后等量混合构建为Mu-F2、Mu-QB、WT-F2、WT-QB 4个混池。然后送至杭州联川生物技术股份有限公司进行样品检测、文库构建、使用Illumina HiSeq 2500平台进行测序,测序深度为30×。
-
通过测序,得到最初的原始序列即Raw Reads。该数据里面含有带接头的和低质量的reads,通过去除接头、过滤掉reads上难确定的碱基类型占比大于10%的及Q≤10碱基数占整reads 50%以上的低质量reads,将低质量的数据过滤掉后得到Clean Reads。以明恢63(MH63RS3,大小为396 Mb)作为参考基因组,用BWA软件将测序获得的Reads定位至基因组上,通过比对定位Clean Reads在参考基因组上的位置,统计各样品的测序深度、基因组覆盖度等信息,并进行变异的检测。
将上一步比对到的结果使用samtools继续过滤掉冗余reads,然后使用GATK对SNP和InDel进行变异检测,4个样本各自生成gVCF后,再进行群体间的基因型比对过滤,最终可以得到变异的位点集。将以上获得的位点使用SnpEff进行变异注释和预测变异影响。
-
在开展关联分析之前,先对SNP和InDel进行筛选,筛选条件包括:剔除具有多个基因型的位点;去除reads支持度低于4的位点;排除混池间基因型相同的位点及隐性混池基因不是来源于隐性亲本的SNP位点。
将经上述条件过滤后的变异位点进行关联分析,有2种方法分别为欧氏距离(euclidean distance, ED)算法和Index算法[13]。ED算法是利用测序数据寻找混池间存在显著差异标记,并以此评估与性状关联区域的方法。理论上,BSA项目构建的2个混池间除了目标性状相关位点存在差异,其他位点均趋向于一致,因此,非目标位点的ED值应趋向于0。ED值越大表明该标记在两混池间的差异越大[22]。SNP-index近年来发表的1种通过混池间的基因型频率差异进行标记关联分析方法,主要是寻找混池之间基因型频率的显著差异。利用2亲本的SNP数据,分别计算2混池的SNP-index,并通过ΔSNP-index观测可能与性状分离相关的位点。通过以上2种算法分别对获得的SNP和InDel分别进行关联分析,得到候选区域。
-
通过将得到的SNP候选区域和InDel候选区域结果取交集,得到最终的候选区域。应用BLAST软件对候选区间内的编码基因进行多个数据库(NR、Swiss-Prot、GO、KEGG、COG)的深度注释。通过详细的注释,快速筛选候选基因。
-
在拔节期取LMM43和WT的叶片,使用天根生化科技有限公司的总RNA提取试剂盒(DP450)提取水稻叶片的RNA,送至公司进行转录组测序。
-
对WT和LMM43突变体进行全生育期观察,发现WT和LMM43在四叶期以前没有出现差异,从四叶期开始LMM43突变体开始在叶尖部位出现少量红褐色的坏死斑点(图1−A)。随着叶片的生长,类病斑斑点数目逐渐增加从叶尖部位逐渐扩散至整个叶片及茎秆(图1−C—E),从幼苗到成熟都带有类病斑表型,类病斑在叶片上随机分布,为扩散的全生育期型突变体。通过对比剑叶、倒二叶、倒三叶发现类病斑在老叶先产生,随后在新叶产生。与WT相比,LMM43突变体叶片枯死,严重时导致整株植株死亡,表明类病斑严重影响了水稻的生长发育(图1)。

图 1 WT和LMM43表型比较
Figure 1. Comparison of phenotypes between WT and LMM43
-
为了确定该突变体的遗传特性,本研究将野生型‘HHZ’作为母本, LMM43为父本进行杂交,通过观察F1表型发现其均表现出类病斑,这说明了LMM43的类病斑性状受到显性基因控制。同时,对F1进行自交后得到的F2代水稻进行了观察,发现类病斑表型在F2代群体中出现了分化,经统计,有斑单株与无斑单株分别为289株和86株,经过卡方检验(χ2=0.854<χ20.05=3.84,df=1),其分离比符合3∶1(表1)。综上确定了LMM43突变体表型是由单个核基因控制的显性性状。
表 1 突变体LMM43和野生型‘HHZ’杂交构建的F2代群体植株表型分离情况
Table 1. Phenotypic segregation of F2 generation population plants constructed from crosses between mutant LMM43 and wild type ‘HHZ’
表型
Phenotype植株数量/株 Number of plants (O-E)2/E 观测值
Observe value理论值
Expected valueO-E 野生型 Wild Type 86 93.75 −7.75 0.641 突变型 Mutants 289 281.25 7.75 0.214 总计 Total 375 375.00 0 0.854 -
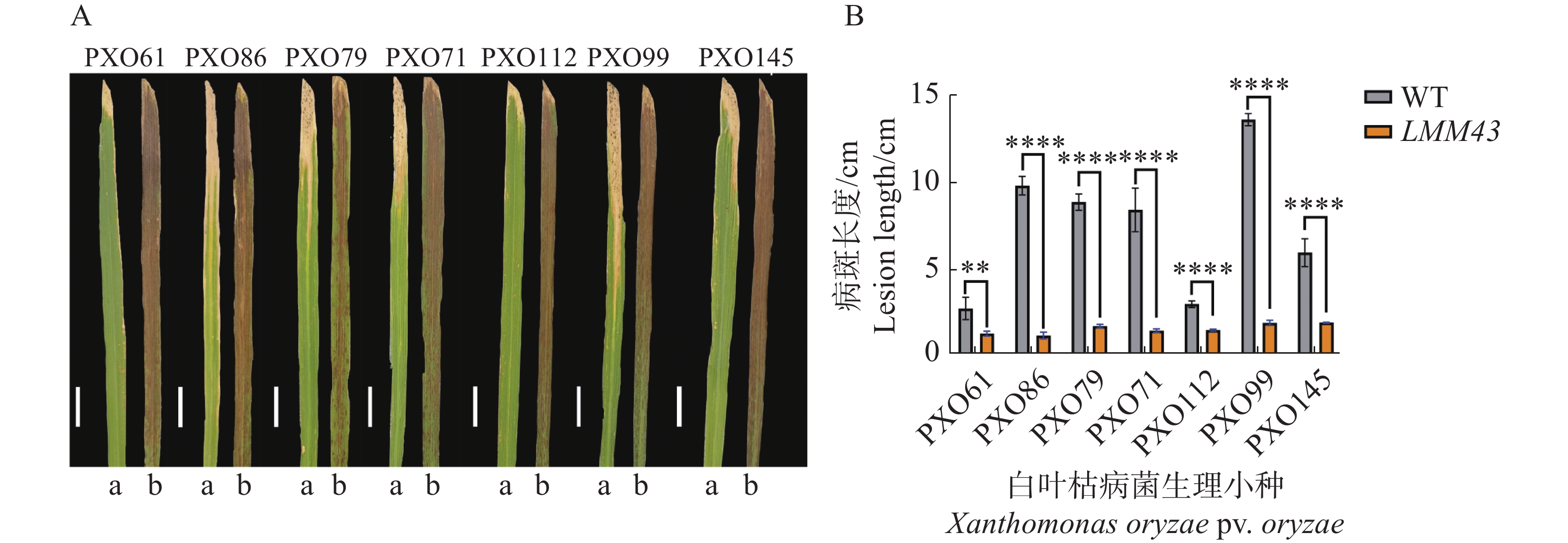
对LMM43突变体进行白叶枯病菌的抗性鉴定,通过接种实验,发现突变体对白叶枯病菌的7个生理小种的抗病性均显著增强(图2)。
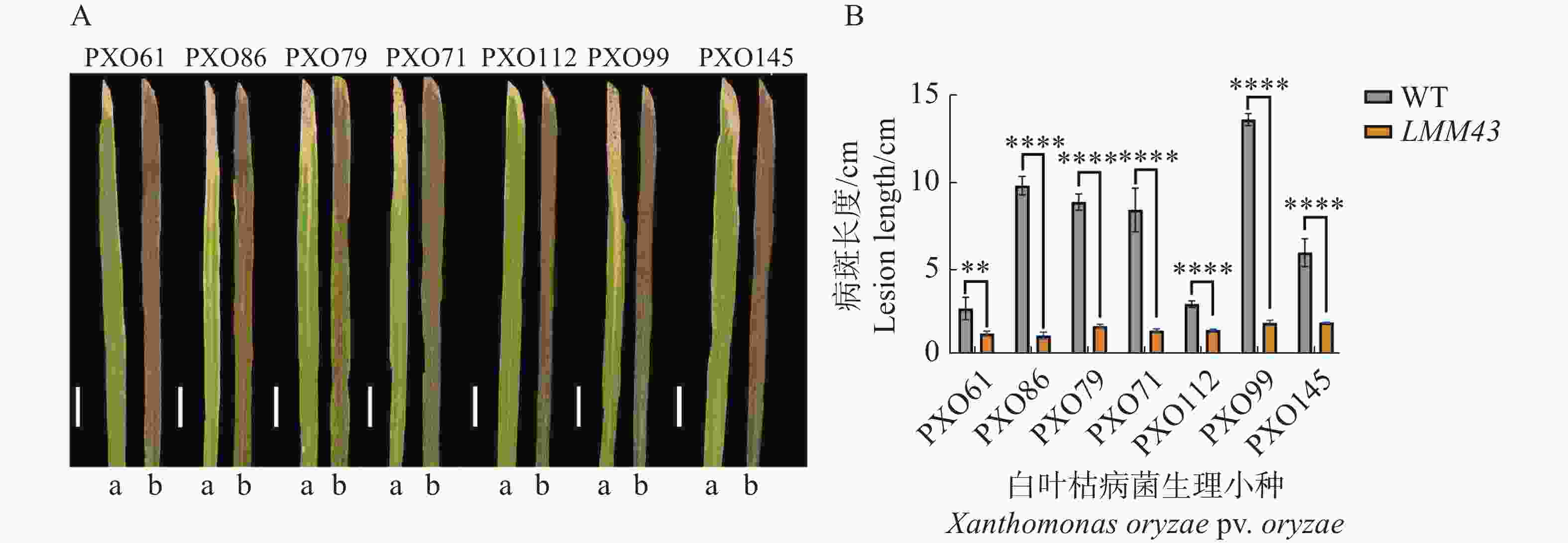
图 2 WT和LMM43白叶枯病菌的抗性鉴定
Figure 2. Resistance characterization of WT and LMM43 leaf blight fungi
-
为了定位引起LMM43表型的基因,分别构建F2类病斑表型(Mu-F2)、LMM43亲本表型(Mu-QB)、F2野生型(WT-F2)、HHZ亲本(WT-QB)4个混池,通过BSA-seq重测序的方法,挖掘造成水稻类病斑突变体表型的基因位点。
-
使用Illumina HiSeq 2500测序平台进行双端测序, 经Raw Data过滤质控后共获得Clean Data为64.55 Gbp,Mu-F2、Mu-QB、WT-F2、WT-QB分别获得了16.32、18.40、16.90、12.93 Gbp的Clean Base,Q30平均值达到了90.96%。将得到的Clean Reads比对至MH63基因组上后,Mu-F2、Mu-QB、WT-F2、WT-QB样本池的比对效率分别为97.83%、97.50%、97.44%、97.32%,平均比对效率达97.52%(表2)。4个样本的平均覆盖深度为39×、44×、40×、31×,平均覆盖深度为38.50×(表2)。以上测序质量统计证明了此次测序质量高,一致性好,可以用于后续的生物信息学分析来定位候选基因。
表 2 样品测序数据评估统计
Table 2. Sample sequencing data evaluation statistics
ID Clean_Reads Clean_Base Q30/% Mapped/% Ave_depth Mu-F2 55 381 938 16 323 284 819 91.05 97.83 39 Mu-QB 62 401 501 18 400 186 668 90.97 97.50 44 WT-F2 57 227 645 16 902 635 145 91.02 97.44 40 WT-QB 6 667 274 12 925 983 271 90.81 97.32 31 注:ID:混池样品编号;Clean_Reads:过滤后的reads对数,即read1和read2记为1条reads;Clean_Base:过滤后的碱基数,Clean Reads数乘以序列长度;Q30:质量值大于等于30的碱基占总碱基数的百分比;Mapped:定位到参考基因组的Clean Reads数占所有Clean Reads数的百分比;Ave_depth:样品平均覆盖深度。下同。 Note: ID: pooled sample number; Clean_reads: log of filtered reads, i.e., read1 and read2 are counted as 1 read; Clean_Base: base count after filtering, Clean Reads multiplied by sequence length; Q30: percentage of bases with a quality value greater than or equal to 30 out of the total base count; Mapped : percentage of Clean Reads located in the reference genome out of all Clean Reads; Ave_depth: average depth of coverage for the sample.similarly hereinafter. -
用BAW进行比对,使用GATK和samtools(v1.9)进行检测和过滤,共发现
1 160 877 个SNP。在Mu-QB和WT-QB间检测,获得76 489 个SNP,造成非同义突变的有8 120 个;Mu-F2和WT-F2间检测SNP,获得128 357 个,非同义突变有12 300 个(表3)。在InDel检测中,亲本混池间共检测到20 742 个InDel,而F2代混池间有32 086 个InDel(表4)。表 3 SNP注释结果统计
Table 3. Statistics of SNP annotation results
Type WT-QB VS
Mu-QBWT-F2 VS
Mu-F2Intergenic 8 820 12 998 Intragenic 10 28 Intron 10 640 20 414 Upstream 21 097 36 848 Downstream 19 145 32 044 Utr_5_Prime 327 827 Utr_3_Prime 441 1 423 Splice_Site_Acceptor 52 82 Splice_Site_Donor 40 76 Splice_Site_Region 208 422 Start_Gained 25 61 Start_Lost 32 59 Synonymous_Coding 7 110 10 188 Non_Synonymous_Coding 8 120 12 300 Synonymous_Stop 8 8 Stop_Gained 374 508 Stop_Lost 40 71 Other 0 0 Total 76 489 128 357 注:Type:SNP所在区域或类型;第2~3列分别为亲本间和混池间存在的对应类型的SNP数量。下同。 Note: Type: the region or type of SNP; columns 2−3 show the number of corresponding types of SNP between parents and pools.the same below. 表 4 InDel注释结果统计
Table 4. Statistics of InDel annotation results
Type WT-QB VS
Mu-QBWT-F2 VS
Mu-F2Intergenic 1 865 2 761 Intragenic 18 28 Intron 650 6 842 Upstream 6 383 10 066 Downstream 5 601 8 495 Utr_5_Prime 324 565 Utr_3_Prime 322 600 Splice_Site_Acceptor 12 19 Splice_Site_Donor 14 31 Splice_Site_Region 83 141 Start_Lost 9 11 Frame_Shift 1 133 1 595 Codon_Deletion 190 280 Codon_Insertion 171 291 Codon_Change_Plus_
Codon_Deletion141 196 Codon_Change_Plus_
Codon_Insertion80 119 Stop_Gained 40 38 Stop_Lost 6 8 Other 0 0 Total 20 742 32 086 -
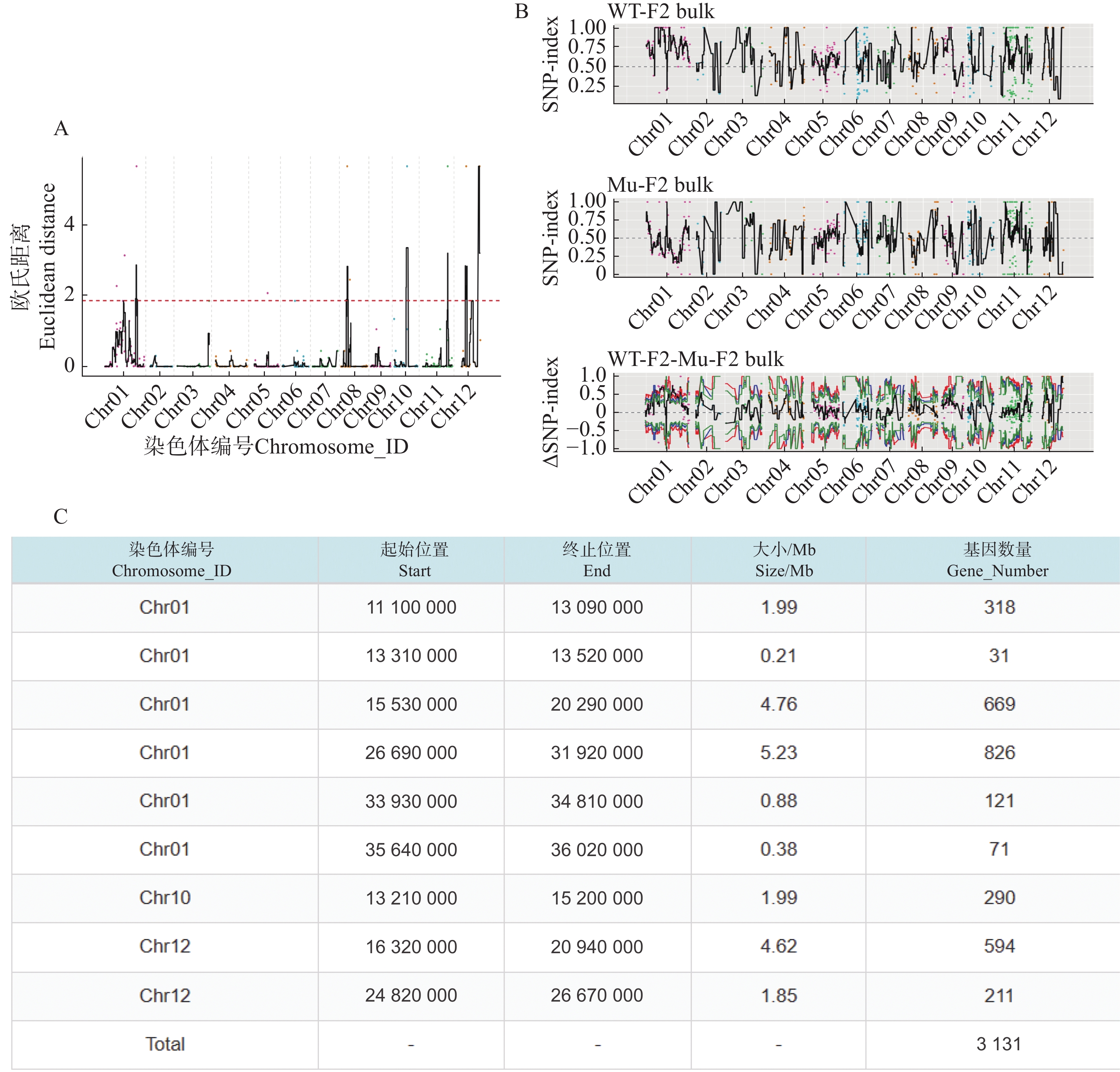
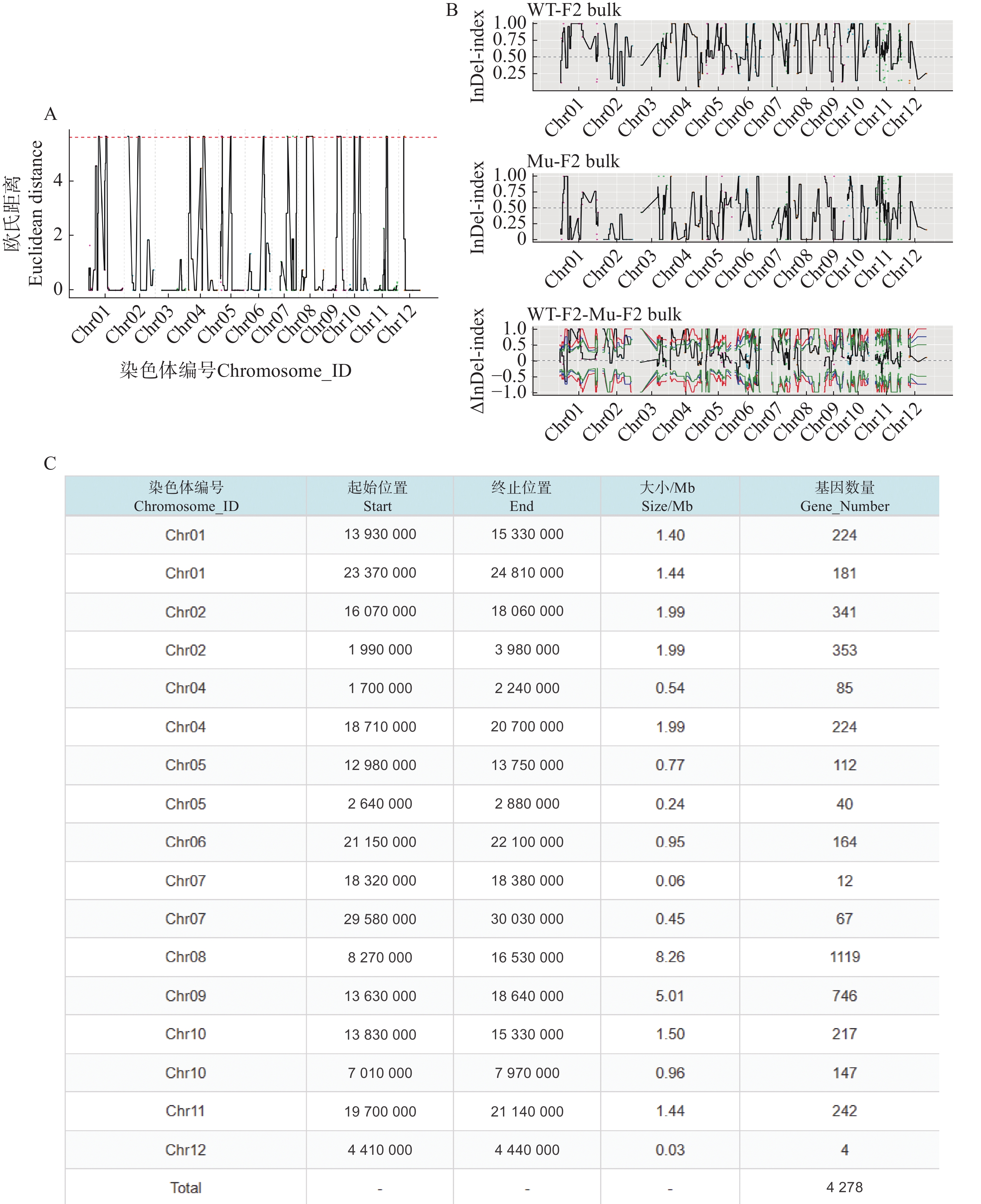
利用F2代的2个混池,将SNP和InDel分别通过ED和Index算法进行关联分析。在ED算法中先计算出每个位点的ED值再进行5次方处理以消除背景噪声,采用Distance法进行拟合后分别可以得到SNP-ED值分布图和InDel-ED值分布图(图3-A、图4−A),然后取所有位点拟合值的median+3SD作为分析的关联阈值(1.86、5.62)即得到8个SNP区域(图3−A中突起峰区域)和17个InDel区域(图4−A中突起峰区域);在Index算法中计算出分别计算出WT-F2和Mu-F2每个位点的SNP-index值,然后两者相减得到ΔSNP-index值,Distance法进行拟合后得到SNP-index值分布图和InDel-index值分布图(图3−B、图4−B),随后取阈值线为置信度0.99,得到了9个SNP区域(图3−B ΔSNP-index值分布图中位于置信度0.99的阈值线上的突起峰)和26个InDel区域(图4−B ΔInDel-index值分布图中位于置信度0.99的阈值线上的突起峰)。将SNP经2种算法得到的8个和9个区域取交集后得到3个SNP关联区域572个位点(图3−C),InDel的17个和26个区域取交集后得到17个关联区域4 278个位点(图4−C)。
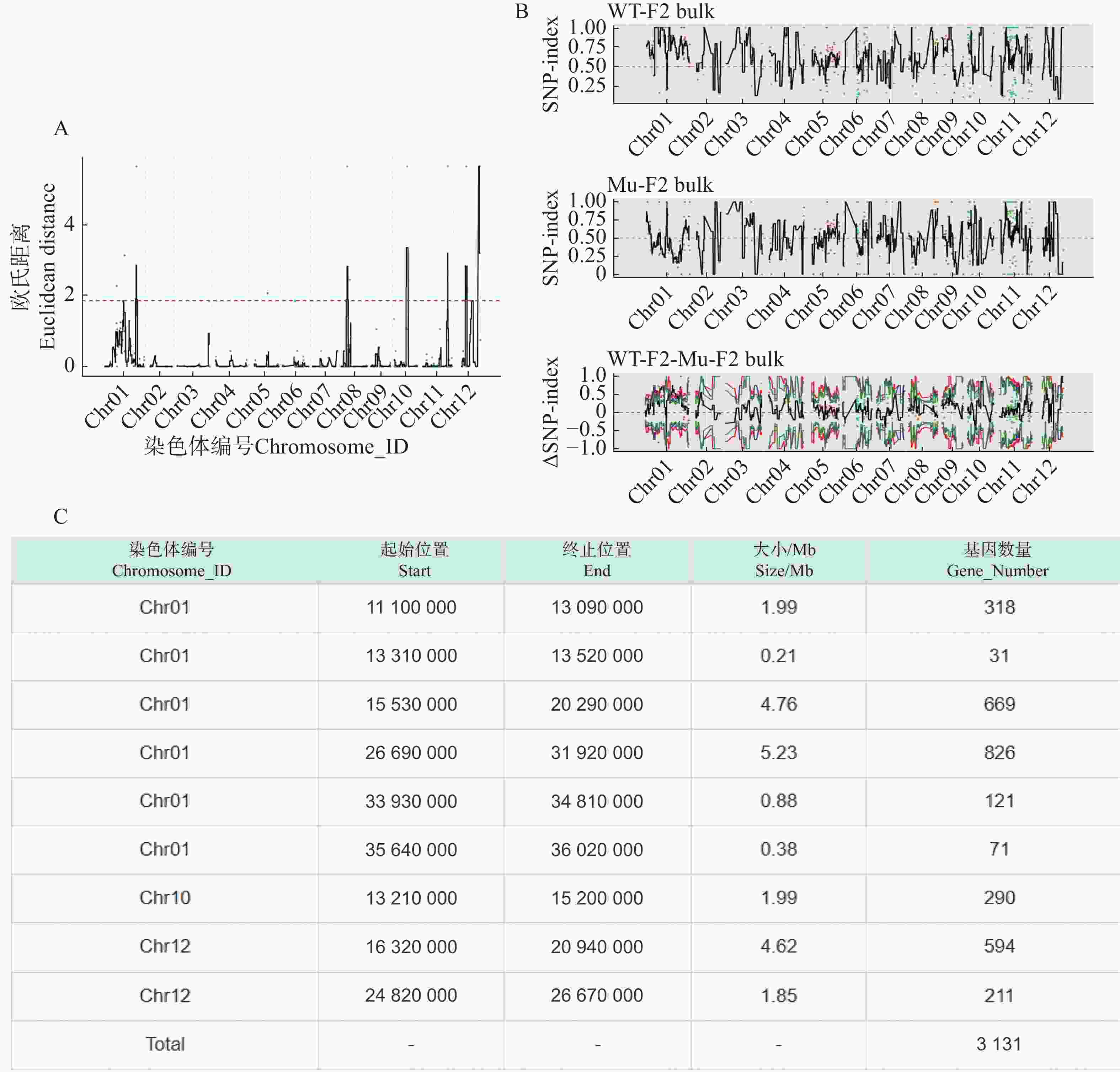
图 3 SNP关联分析图
Figure 3. SNP association analysis graphs
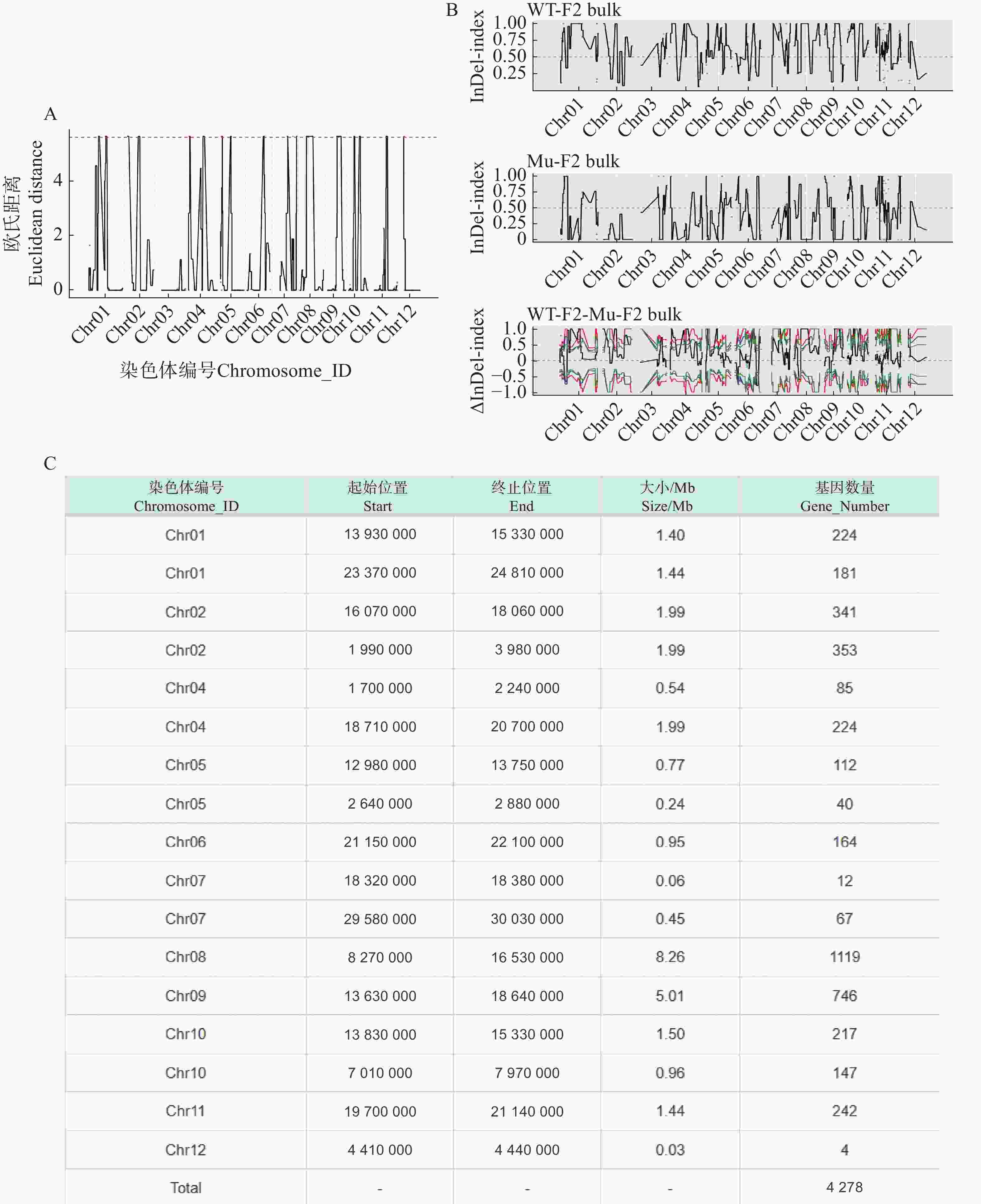
图 4 InDel关联分析图
Figure 4. InDel association analysis graphs
-
根据以上的关联结果再将获得的3个SNP关联区域和17个InDel关联区域取交集后得到了一个候选区域。在10号染色体13 830 000~15 200 000的1.37 Mb内(表5),共199个基因。其中,根据这些变异位点的注释可知,亲本间存在的非同义突变20个,移码突变的InDel 2个;混池间非同义突变26个,移码突变的InDel 2个;所以在该候选区间内可能与性状密切相关的为这46个非同义突变的SNP位点和4个移码突变的InDel位点。通过对候选位点的编码基因进行多个数据库(NR、Swiss-Prot、GO、KEGG、COG)的深度注释,共注释到16个非同义突变基因,3个移码突变基因(表6)。
表 5 最终关联区域信息统计表
Table 5. Statistical table of information on final associated regions
染色体编号
Chromosome_ID起始位置
Start终止位置
End大小/Mb
Size/Mb基因数量
Gene_NumberChr10 13830 00015200000 1.37 199 表 6 候选基因列表
Table 6. List of candidate genes
序号
Number基因登录号
Gene_ID变异类型
Type功能注释数据库
Annotated_Databases1 OsMH_10G0226700 Non_Synonymous_Coding shewanella-like protein phosphatase 1 2 OsMH_10G0225900 Non_Synonymous_Coding Protein TIFY 11d 3 OsMH_10G0224100 Non_Synonymous_Coding protein Strubbelig-Receptor Family7 isoform X1 4 OsMH_10G0223300 Non_Synonymous_Coding red chlorophyll catabolite reductase 5 OsMH_10G0222600 Non_Synonymous_Coding F-box/kelch-repeat protein SKIP30 6 OsMH_10G0221000 Non_Synonymous_Coding ethanolamine-phosphate cytidylyltransferase 7 OsMH_10G0208500 Non_Synonymous_Coding protein Gravitropic In The Light 1 8 OsMH_10G0208400 Non_Synonymous_Coding protein CutA 1, chloroplastic 9 OsMH_10G0208100 Non_Synonymous_Coding pyridoxine/pyridoxamine 5'-phosphate oxidase 1,
chloroplastic isoform X110 OsMH_10G0206700 Non_Synonymous_Coding calmodulin-binding transcription activator 3 isoform X2 11 OsMH_10G0202100 Non_Synonymous_Coding ethylene-responsive transcription factor RAP2-13 12 OsMH_10G0201500 Non_Synonymous_Coding protein NRT1/ PTR FAMILY 8.3 isoform X1 13 OsMH_10G0201300 Non_Synonymous_Coding probable glucan 1,3-beta-glucosidase A 14 OsMH_10G0201200 Non_Synonymous_Coding disease resistance protein RGA4-like 15 OsMH_10G0201000 Non_Synonymous_Coding probable protein phosphatase 2C 71 16 OsMH_10G0200600 Non_Synonymous_Coding scarecrow-like protein 21 17 OsMH_10G0223500 Frame_Shift hypothetical protein 18 OsMH_10G0116300 Frame_Shift uncharacterized protein 19 OsMH_10G0211100 Frame_Shift tyrosine decarboxylase-like -
通过以上综合分析,得到了OsMH_10G0226700、OsMH_10G0225900等19个基因,其中,16个为非同义突变,3个为移码突变,鉴于EMS造成的主要为G到A和C到T的点突变,所以非同义突变是需要重点关注的。在这16个非同义突变中,其中5个基因可能与水稻的类病斑性状有关,包括OsMH_10G0223300, red chlorophyll catabolite reductase(PCCR); OsMH_10G0206700, calmodulin-binding transcription activator 3 isoform X2(CAMTA3); OsMH_10G0202100, ethylene-responsive transcription factor RAP2-13(RAP2-13); OsMH_10G0201300, probable glucan 1,3-beta-glucosidase A(β−1,3−葡聚糖苷酶A); OsMH_10G0201200, disease resistance protein RGA4-like(RGA4-like)(表6)。
-
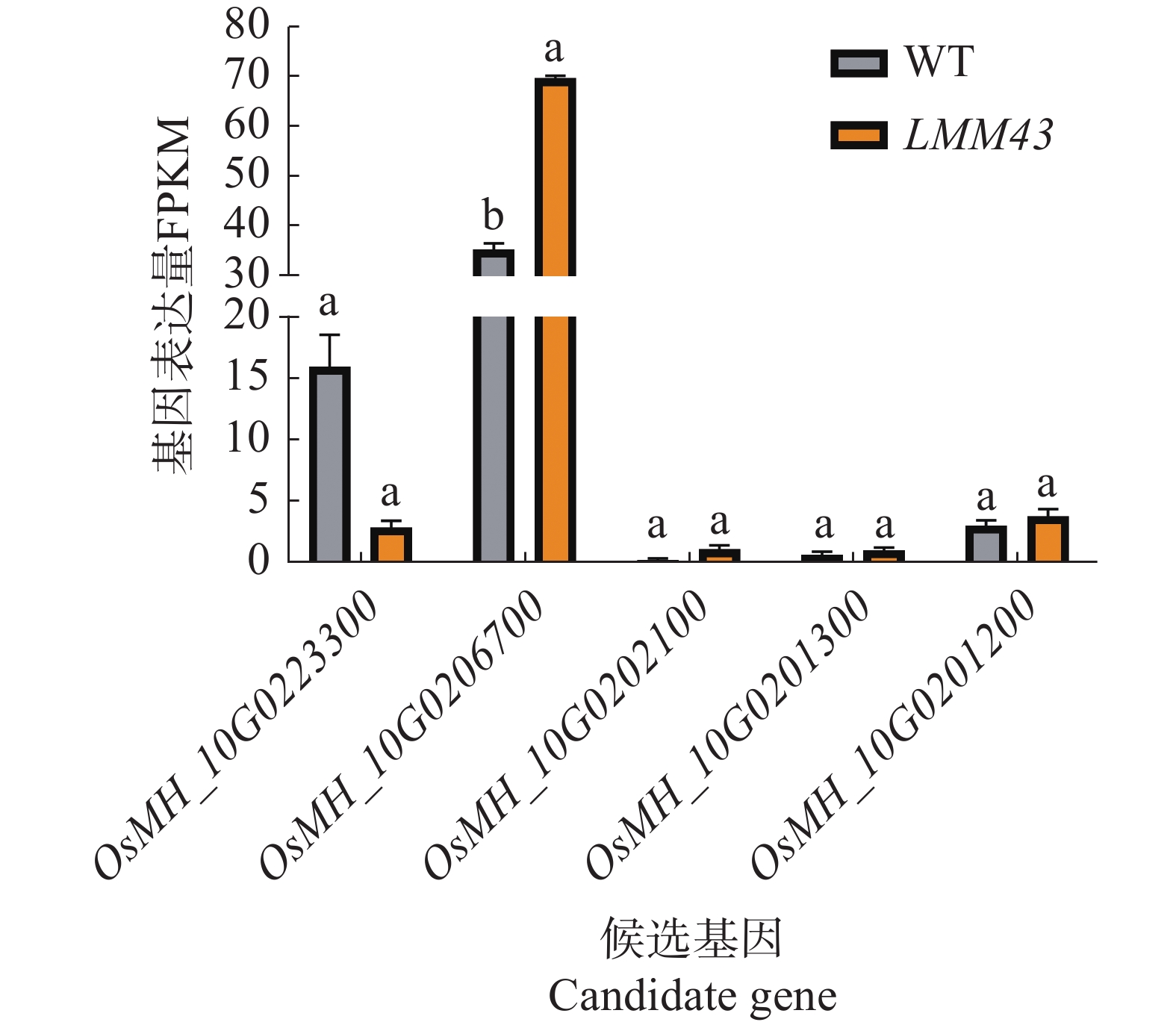
为了解候选基因LMM43突变体中是否有表达量的变化,通过转录组测序结果分析5个候选基因的表达量变化,结果表明,只有OsMH_10G0223300和OsMH_10G0206700在水稻中的基因表达量较高,其中OsMH_10G0223300的表达量下降,但不显著;OsMH_10G0206700的基因表达量在LMM43突变体中显著上调(图5)。
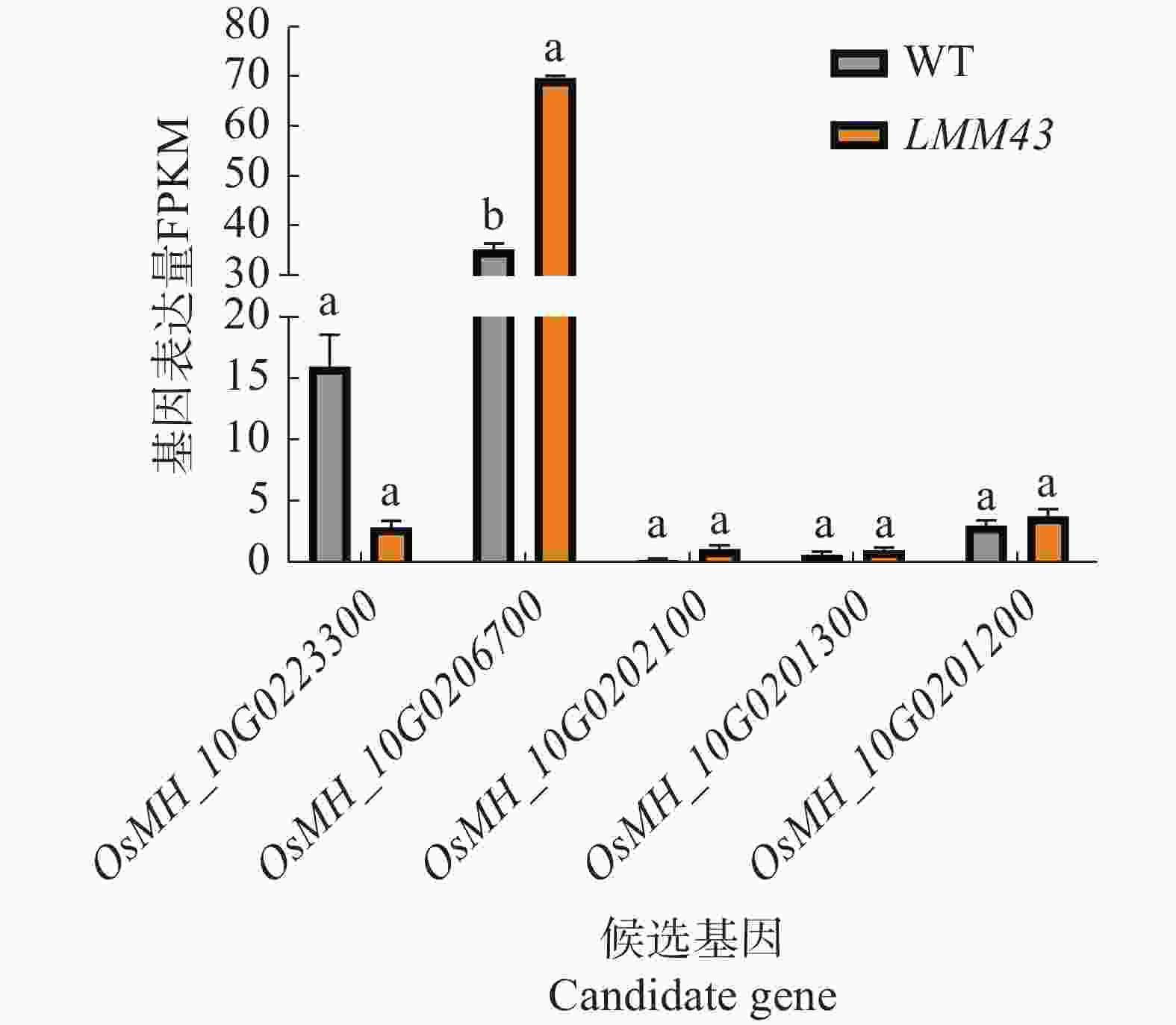
图 5 候选基因表达量分析
Figure 5. Candidate gene expression analysis
-
水稻类病斑自1965年被发现后,到目前为止已有近100多个基因被报道[3]。研究表明,大部分水稻类病斑突变体对白叶枯病、稻瘟病或纹枯病具有抗性。水稻的抗病机制,特别是类病斑突变体的抗病机制,对于提高水稻的抗病性和产量具有重要意义。但是在水稻类病斑形成和激活免疫防御基因高表达的同时,类病斑突变体的生长发育同样受到了严重的影响,如突变体 lms1[23]、spl28[24]等不仅表现出类病斑的表型, 而且植株矮小,存在早衰现象;spl29[25],spl33 [26]等突变体的株高、穗长、结实率和千粒重与野生型相比均显著下降。本研究中的LMM43突变体类病斑属于扩散型的全生育期类病斑突变体,其生长发育和已报道过的类病斑突变体一样,类病斑的大面积的蔓延也会使得其农艺性状的下降,同时植株的矮缩也让LMM43的结实较为困难。
第1个突变体sl[27],是自然变异引起的,后来随着科学技术手段的发展,人们通过物理、化学及生物诱变型的多种手段获得突变体。目前已克隆近50个水稻类病斑突变体基因,其中大多数是通过图位克隆获得的[28],其中大部分为单个核基因控制隐性遗传,极少数的如显性基因遗传突变体有Spl18[7]、spl-D[9]、Spl26[10],半显性遗传突变体LIL1[29]、nls1-1D[8]。其中,nls1-1D类病斑突变纯合体后代在生长方面存在着严重的缺陷,发芽后3周内就会死亡。所以显性水稻类病斑突变体需要构建F2:3群体使用纯合体进行定位,在F2代杂合基因型干扰及纯合后代材料的致死现象,使得基因定位上存在着较大困难。
BSA-seq是一种结合了分离体混合分析和高通量测序技术的方法,可以用于快速定位与特定性状相关的基因,该技术的核心在于选择具有目标性状极端表型的个体[13]。所以选择了SBA-seq来对LMM43类病斑突变体的基因进行初步定位,由于使用F2代群体构建极端性状混池,其中,有显性杂合体的存在,使得干扰较多,最终通过分析,将候选区域确定在了10号染色体的1.37 Mb的一个区间内,包括199个基因。根据目前报道过已经克隆的10号染色体上共有5个类病斑突变体,其中4个隐性突变,1个显性突变,都不在本研究的候选区域内[3],证明了LMM43是由一个尚未报道全新的显性基因控制的类病斑突变体。在此基础上通过经候选区域内的基因经多个数据库深度注释后确定了5个候选基因。OsMH_10G0223300编码的PCCR,是植物中叶绿素降解的关键酶,Tang等[30]发现敲除OsPCCR1会使水稻产生类病斑;OsMH_10G0206700编码的CAMTA3,是一类转录因子可感知来自Ca2+的应激信号,具有多种胁迫响应性[31];OsMH_10G0202100编码的RAP2-13,作为乙烯响应因子(ERF)家族,可能参与了水稻的抗病等过程;OsMH_10G0201300编码的β−1,3−葡聚糖苷酶A主要参与病原体防御、碳水化合物代谢和逆境响应等过程[32];OsMH_10G0201200编码的RGA4-like,是RGA4的同源蛋白,Zhu等[33]在研究中发现了RGA4在水稻中通过与RGA5协同作用,识别稻瘟菌的效应蛋白并激活抗病反应。综上可知,这些基因可能与类病斑性状的出现相关。通过对候选基因的表达量进行分析发现只有2个基因在水稻中的表达量较高,OsMH_10G0223300的表达量下降,OsMH_10G0206700显著上调,由于碱基突变造成的非同义突变不一定会影响该基因的表达量变化,所以后续需要继续通过互补实验进行验证。
本研究从籼稻‘黄华占’(‘HHZ’)经EMS诱变后的群体中分离了1个水稻类病斑突变体LMM43,通过对LMM43类病斑突变体的表型进行全生育期的观察,是1个全生育期扩散型的类病斑突变体,且LMM43突变体对白叶枯病菌的抗性显著增强。在F2代群体中,通过统计性状分离情况,得知LMM43突变体表型是由单个核基因控制的显性性状。利用BSA-seq对LMM43类病斑突变体的基因进行初步定位。在‘HHZ’×LMM43杂交构建的F2代分离群体中进行混池构建,将基因定位在水稻10号染色体的1个约1.37 Mb的区间内,该区间包含199个预测基因,综合分析预测出5个候选基因,为后期基因克隆和功能研究奠定了基础。
基于BSA-seq对一个水稻类病斑基因的初步定位
DOI: 10.15886/j.cnki.rdswxb.20250022
 CSTR: 32425.14.j.cnki.rdswxb.20250022
CSTR: 32425.14.j.cnki.rdswxb.20250022
Preliminary localization of a rice lesion mimic gene based on BSA-seq
-
摘要: 类病斑突变体(lesion mimic mutants, LMM)在植物免疫和生长发育研究中具有重要意义。本研究旨在通过BSA-seq(bulked segregant analysis sequencing)技术对1个水稻类病斑基因进行初步定位。首先,从籼稻(Oryza sativa ssp.) 品种‘黄华占’(‘HHZ’)经EMS诱变后的群体中筛选出1个出现类病斑症状的突变体LMM43,通过LMM43与野生型亲本杂交后得到的F2代分离群体进行表型分析,确定类病斑性状的遗传模式。利用BSA-seq技术,对类病斑表型的个体和正常个体的DNA混合样本进行高通量测序。通过关联分析单核苷酸多态性(single nucleotide polymorphism, SNP)及插入缺失(insertion and deletion, InDel)的差异结合表现型分析,将目标基因初步定位在水稻第10号染色体的一段区间内,该区间长度约为1.37 Mb,包含199个基因,最终确定了5个候选基因。Abstract: Lesion mimic mutants (LMM) are important in plant immunity and growth and development studies. The aim of this study was to perform a preliminary localization of a rice lesion mimic gene by BSA-seq (Bulked Segregant Analysis sequencing) technique. First, we screened a mutant, LMM43, from the population of indica rice cultivar Huang Huazhan (HHZ) mutagenized by EMS for the appearance of lesion-like symptoms, and phenotyped the F2 segregant population obtained by crossing LMM43 with the wild-type parent to determine the mode of inheritance of the lesion-like trait. High-throughput sequencing was performed on mixed samples of DNA from individuals with disease-like spot phenotypes and normal individuals using BSA-seq technology. Through the association analysis of Single Nucleotide Polymorphism (SNP) and Insertion and Deletion (InDel) differences combined with phenotypic analysis, the target genes were preliminarily localized in a segment of rice chromosome 10, which is about 1.37 Mb in length and contains 199 genes. The length of this interval is about 1.37 Mb, which contains 199 genes, and five candidate genes were finally identified. The preliminary localization results laid the foundation for further gene cloning and functional verification, and helped to deeply understand the molecular mechanism of rice-like spot mutants, which is of great theoretical and applied value for the study of rice disease resistance breeding and plant immunity mechanism.
-
Key words:
- Rice /
- Lesion mimic /
- Disease resistance /
- BSA-seq
-
图 2 WT和LMM43白叶枯病菌的抗性鉴定
A. 白叶枯病菌7个生理小种接菌20 d的病斑特征;B. 接菌20 d的病斑长度(cm),a为WT, b为LMM43。标尺.A图2 cm。数据来源于3个重复,误差棒为3个重复的平均数±标准差,采用t-检验,**表示P<0.01,****表示P<0.000 1。
Fig. 2 Resistance characterization of WT and LMM43 leaf blight fungi
A. Characterization of spots inoculated with seven physiological mini-inocula of leaf blight bacteria for 20 d. B. Spot lengths(cm)at 20 d of inoculation, a for HHZ, b for LMM43. Scale bar. 2 cm in Fig. A. The data are from 3 replicates, and error bars are the mean±standard deviation of the 3 replicates using the t-test, ** denotes P<0.01, **** denotes P<0.000 1.
图 3 SNP关联分析图
A. SNP-ED值分布图;B. SNP-index值分布图;C. SNP通过2种算法关联区域信息统计表。
Fig. 3 SNP association analysis graphs
A. Distribution of SNP-ED values; B.Distribution of SNP-index values; C. Statistical table of SNP association region information by 2 algorithms.
图 4 InDel关联分析图
A. InDel-ED值分布图;B. InDel-index值分布图;C. InDel通过2种算法关联区域信息统计表。
Fig. 4 InDel association analysis graphs
A. Distribution of InDel-ED values; B. Distribution of InDel-index values; C. Statistical table of InDel association region information by 2 algorithms.
图 5 候选基因表达量分析
数据来源于3个重复,误差棒为3个重复的平均数±标准差,以Fold-Change ≥ 2且FDR < 0.05检验显著性,不同小写字母表示基因表达差异具有显著性变化。
Fig. 5 Candidate gene expression analysis
Data from 3 replicates, error bars are mean ± standard deviation of 3 replicates, significance is tested by Fold-Change ≥ 2 and FDR < 0.05, and different lowercase letters indicate significant changes in gene expression differences.
表 1 突变体LMM43和野生型‘HHZ’杂交构建的F2代群体植株表型分离情况
Table 1 Phenotypic segregation of F2 generation population plants constructed from crosses between mutant LMM43 and wild type ‘HHZ’
表型
Phenotype植株数量/株 Number of plants (O-E)2/E 观测值
Observe value理论值
Expected valueO-E 野生型 Wild Type 86 93.75 −7.75 0.641 突变型 Mutants 289 281.25 7.75 0.214 总计 Total 375 375.00 0 0.854  下载: 导出CSV
下载: 导出CSV
表 2 样品测序数据评估统计
Table 2 Sample sequencing data evaluation statistics
ID Clean_Reads Clean_Base Q30/% Mapped/% Ave_depth Mu-F2 55 381 938 16 323 284 819 91.05 97.83 39 Mu-QB 62 401 501 18 400 186 668 90.97 97.50 44 WT-F2 57 227 645 16 902 635 145 91.02 97.44 40 WT-QB 6 667 274 12 925 983 271 90.81 97.32 31 注:ID:混池样品编号;Clean_Reads:过滤后的reads对数,即read1和read2记为1条reads;Clean_Base:过滤后的碱基数,Clean Reads数乘以序列长度;Q30:质量值大于等于30的碱基占总碱基数的百分比;Mapped:定位到参考基因组的Clean Reads数占所有Clean Reads数的百分比;Ave_depth:样品平均覆盖深度。下同。 Note: ID: pooled sample number; Clean_reads: log of filtered reads, i.e., read1 and read2 are counted as 1 read; Clean_Base: base count after filtering, Clean Reads multiplied by sequence length; Q30: percentage of bases with a quality value greater than or equal to 30 out of the total base count; Mapped : percentage of Clean Reads located in the reference genome out of all Clean Reads; Ave_depth: average depth of coverage for the sample.similarly hereinafter.
下载: 导出CSV
表 3 SNP注释结果统计
Table 3 Statistics of SNP annotation results
Type WT-QB VS
Mu-QBWT-F2 VS
Mu-F2Intergenic 8 820 12 998 Intragenic 10 28 Intron 10 640 20 414 Upstream 21 097 36 848 Downstream 19 145 32 044 Utr_5_Prime 327 827 Utr_3_Prime 441 1 423 Splice_Site_Acceptor 52 82 Splice_Site_Donor 40 76 Splice_Site_Region 208 422 Start_Gained 25 61 Start_Lost 32 59 Synonymous_Coding 7 110 10 188 Non_Synonymous_Coding 8 120 12 300 Synonymous_Stop 8 8 Stop_Gained 374 508 Stop_Lost 40 71 Other 0 0 Total 76 489 128 357 注:Type:SNP所在区域或类型;第2~3列分别为亲本间和混池间存在的对应类型的SNP数量。下同。 Note: Type: the region or type of SNP; columns 2−3 show the number of corresponding types of SNP between parents and pools.the same below.
下载: 导出CSV
表 4 InDel注释结果统计
Table 4 Statistics of InDel annotation results
Type WT-QB VS
Mu-QBWT-F2 VS
Mu-F2Intergenic 1 865 2 761 Intragenic 18 28 Intron 650 6 842 Upstream 6 383 10 066 Downstream 5 601 8 495 Utr_5_Prime 324 565 Utr_3_Prime 322 600 Splice_Site_Acceptor 12 19 Splice_Site_Donor 14 31 Splice_Site_Region 83 141 Start_Lost 9 11 Frame_Shift 1 133 1 595 Codon_Deletion 190 280 Codon_Insertion 171 291 Codon_Change_Plus_
Codon_Deletion141 196 Codon_Change_Plus_
Codon_Insertion80 119 Stop_Gained 40 38 Stop_Lost 6 8 Other 0 0 Total 20 742 32 086
下载: 导出CSV
表 5 最终关联区域信息统计表
Table 5 Statistical table of information on final associated regions
染色体编号
Chromosome_ID起始位置
Start终止位置
End大小/Mb
Size/Mb基因数量
Gene_NumberChr10 13830 00015200000 1.37 199
下载: 导出CSV
表 6 候选基因列表
Table 6 List of candidate genes
序号
Number基因登录号
Gene_ID变异类型
Type功能注释数据库
Annotated_Databases1 OsMH_10G0226700 Non_Synonymous_Coding shewanella-like protein phosphatase 1 2 OsMH_10G0225900 Non_Synonymous_Coding Protein TIFY 11d 3 OsMH_10G0224100 Non_Synonymous_Coding protein Strubbelig-Receptor Family7 isoform X1 4 OsMH_10G0223300 Non_Synonymous_Coding red chlorophyll catabolite reductase 5 OsMH_10G0222600 Non_Synonymous_Coding F-box/kelch-repeat protein SKIP30 6 OsMH_10G0221000 Non_Synonymous_Coding ethanolamine-phosphate cytidylyltransferase 7 OsMH_10G0208500 Non_Synonymous_Coding protein Gravitropic In The Light 1 8 OsMH_10G0208400 Non_Synonymous_Coding protein CutA 1, chloroplastic 9 OsMH_10G0208100 Non_Synonymous_Coding pyridoxine/pyridoxamine 5'-phosphate oxidase 1,
chloroplastic isoform X110 OsMH_10G0206700 Non_Synonymous_Coding calmodulin-binding transcription activator 3 isoform X2 11 OsMH_10G0202100 Non_Synonymous_Coding ethylene-responsive transcription factor RAP2-13 12 OsMH_10G0201500 Non_Synonymous_Coding protein NRT1/ PTR FAMILY 8.3 isoform X1 13 OsMH_10G0201300 Non_Synonymous_Coding probable glucan 1,3-beta-glucosidase A 14 OsMH_10G0201200 Non_Synonymous_Coding disease resistance protein RGA4-like 15 OsMH_10G0201000 Non_Synonymous_Coding probable protein phosphatase 2C 71 16 OsMH_10G0200600 Non_Synonymous_Coding scarecrow-like protein 21 17 OsMH_10G0223500 Frame_Shift hypothetical protein 18 OsMH_10G0116300 Frame_Shift uncharacterized protein 19 OsMH_10G0211100 Frame_Shift tyrosine decarboxylase-like
下载: 导出CSV
-
[1] 廖海丽, 刘剑镔, 梁毅, 等. 水稻类病斑突变体及其发生机制研究进展 [J]. 杂交水稻, 2024, 39(5): 1−12. [2] 张宇,王金开,陈小林, 等. 水稻响应白叶枯病菌侵染的转录组分析[J]. 南方农业学报, 2023, 54(3): 815 − 828. [3] 林祯芃, 曾维, 郭铧艳, 等. 水稻类病斑相关基因的克隆及调控机制研究进展[J/OL]. 分子植物育种,1−13 (2022-10-17)[2025-01-20]https://kns.cnki.net/kcms/detail/46.1068.S.20221014.1637.014.html. [4] FEKIH R, TAMIRU M, KANZAKI H, et al. The rice (Oryza sativa l.) lesion mimic resembling, which encodes an aaa-type ATPase, is implicated in defense response[J]. Molecular Genetics and Genomics, 2015, 290(2): 611 − 622. doi: 10.1007/s00438-014-0944-z [5] XU X, CHEN Z, SHI Y F, et al. Functional inactivation of OsGCNT induces enhanced disease resistance to Xanthomonas oryzae pv. oryzae in rice[J]. BMC Plant Biology, 2018, 18(1): 264. doi: 10.1186/s12870-018-1489-9 [6] BAI W, WANG P, HONG J, et al. Earlier degraded Tapetum1 (EDT1) encodes an ATP-citrate lyase required for tapetum programmed cell death[J]. Plant Physiology, 2019, 181(3): 1223 − 1238. doi: 10.1104/pp.19.00202 [7] MORI M, TOMITA C, SUGIMOTO K, et al. Isolation and molecular characterization of a Spotted leaf 18 mutant by modified activation-tagging in rice[J]. Plant Molecular Biology, 2007, 63(6): 847 − 860. doi: 10.1007/s11103-006-9130-y [8] TANG J, ZHU X, WANG Y, et al. Semi-dominant mutations in the CC-NB-LRR-type R gene, NLS1 lead to constitutive activation of defense responses in rice[J]. The Plant Journal, 2011, 66(6): 996 − 1007. doi: 10.1111/j.1365-313X.2011.04557.x [9] ZHAO X, QIU T, FENG H, et al. A novel Glycine-rich domain protein, GRDP1, functions as a critical feedback regulator for controlling cell death and disease resistance in rice[J]. Journal of Experimental Botany, 2021, 72(2): 608 − 622. doi: 10.1093/jxb/eraa450 [10] SHANG H, LI P, ZHANG X, et al. The gain-of-function mutation, OsSpl26, positively regulates plant immunity in rice[J]. International Journal of Molecular Sciences, 2022, 23(22): 14168. doi: 10.3390/ijms232214168 [11] CAI L, YAN M, YUN H, et al. Identification and fine mapping of lesion mimic mutant spl36 in rice (Oryza sativa L.)[J]. Breeding Science, 2021, 71(5): 510 − 519. doi: 10.1270/jsbbs.20160 [12] KANG S G, LEE K E, SINGH M, et al. Rice lesion mimic mutants (LMM): the current understanding of genetic mutations in the failure of ROS scavenging during lesion formation[J]. Plants, 2021, 10(8): 1598. doi: 10.3390/plants10081598 [13] 张刚, 朱林, 聂豪杰, 等. 基于文献计量学BSA在作物育种领域的应用现状与展望[J]. 遗传, 2024, 46(5): 360 − 372. [14] 林秋云, 王克喜, 胡伟, 等. 水稻矮秆多分蘖突变体st1的表型特征与遗传分析[J]. 江苏农业科学, 2024, 52(12): 75 − 79. [15] ABE A, KOSUGI S, YOSHIDA K, et al. Genome sequencing reveals agronomically important loci in rice using MutMap[J]. Nature Biotechnology, 2012, 30(2): 174 − 178. doi: 10.1038/nbt.2095 [16] 张艳萍, 叶春雷, 齐燕妮, 等. 基于BSA-Seq定位胡麻耐盐相关基因位点[J]. 北方园艺, 2025(3): 27 − 34. doi: 10.11937/bfyy.20242807 [17] 陈丽, 孙建昌, 王昕. 基于BSA-seq法的水稻稻瘟病抗性基因定位[J]. 中国稻米, 2024, 30(6): 35 − 41. doi: 10.3969/j.issn.1006-8082.2024.06.006 [18] 魏荣华, 尹明, 王文生, 等. 基于BSA-seq发掘水稻抽穗期相关QTLs及候选基因[J]. 中国农业科技导报, 2024, 26(9): 12 − 24. [19] 杜灿灿, 曾生元, 景德道, 等. 基于BSA-seq的一个苗期黄化转绿突变体基因定位[J]. 江苏农业科学, 2024, 52(12): 53 − 60. [20] 徐乾坤. 水稻类病斑基因LML11的图位克隆与功能分析 [D]. 重庆: 西南大学, 2021. [21] MANOSALVA P M, BRUCE M, LEACH J E. Rice 14-3-3 protein (GF14e) negatively affects cell death and disease resistance[J]. The Plant Journal, 2011, 68(5): 777 − 787. doi: 10.1111/j.1365-313X.2011.04728.x [22] 孙天宇. 大豆开花期相关数量性状位点(QTL)的定位[D]. 北京: 中国科学院大学(中国科学院东北地理与农业生态研究所), 2017. [23] 夏赛赛, 崔玉, 李凤菲, 等. 水稻类病斑早衰突变体lmps1的表型鉴定与基因定位[J]. 作物学报, 2018, 45(1): 46 − 54. [24] QIAO Y, JIANG W, LEE J, et al. SPL28 encodes a clathrin-associated adaptor protein complex 1, medium subunit μ1 (AP1M1) and is responsible for spotted leaf and early senescence in rice (oryza sativa)[J]. New Phytologist, 2010, 185(1): 258 − 274. doi: 10.1111/j.1469-8137.2009.03047.x [25] WANG Z, WANG Y, HONG X, et al. Functional inactivation of UDP-N-acetylglucosamine pyrophosphorylase 1 (UAP1) induces early leaf senescence and defence responses in rice[J]. Journal of Experimental Botany, 2015, 66(3): 973 − 987. doi: 10.1093/jxb/eru456 [26] WANG S, LEI C, WANG J, et al. SPL33, encoding an eEF1A-like protein, negatively regulates cell death and defense responses in rice[J]. Journal of Experimental Botany, 2017, 68(5): 899 − 913. doi: 10.1093/jxb/erx001 [27] TAKAHASHI A, KAWASAKI T, HENMI K, et al. Lesion mimic mutants of rice with alterations in early signaling events of defense[J]. The Plant Journal: for Cell and Molecular Biology, 1999, 17(5): 535 − 545. doi: 10.1046/j.1365-313X.1999.00405.x [28] YIN W, ZHONG Q, ZHU Z, et al. LMI1 a DUF292 protein family gene, regulates immune responses and cell death in rice[J]. The Crop Journal, 2024, 12(6): 1619 − 1632. doi: 10.1016/j.cj.2024.07.015 [29] ZHOU Q, ZHANG Z, LIU T, et al. Identification and map-based cloning of the light-induced lesion mimic mutant 1 (LIL1) gene in rice[J]. Frontiers in Plant Science, 2017, 8: 2122. doi: 10.3389/fpls.2017.02122 [30] TANG Y, LI M, CHEN Y, et al. Knockdown of OsPAO and OsRCCR1 cause different plant death phenotypes in rice[J]. Journal of Plant Physiology, 2011, 168(16): 1952 − 1959. doi: 10.1016/j.jplph.2011.05.026 [31] NOMAN M, AYSHA J, KETEHOULI T, et al. Calmodulin binding transcription activators: an interplay between calcium signalling and plant stress tolerance[J]. Journal of Plant Physiology, 2021, 256: 153327. doi: 10.1016/j.jplph.2020.153327 [32] 万凌琳. 水稻β-1, 3葡聚糖苷酶基因Osg1的功能研究 [D]. 武汉: 武汉大学, 2010. [33] ZHU T, WU X, YUAN G, et al. A resurfaced sensor NLR confers new recognition specificity to non-MAX effectors[J]. Journal of Integrative Plant Biology, 2025, 67(1): 11 − 14. doi: 10.1111/jipb.13805 -
 点击查看大图
点击查看大图

计量
- 文章访问数: 1414
- HTML全文浏览量: 796
- PDF下载量: 43
- 被引次数: 0
