

-
白边侧足海天牛(Elysia leucolegnote)属于软体动物门囊舌目(Sacoglossa)海天牛超科(Elysioidea)海天牛属(Elysia),该种分布于中国的香港[1]、海口[2]以及泰国[3]、菲律宾[4]等地红树林。目前,在我国已报道有8种海天牛的分布记录[1,5]。海天牛属部分种具有保留藻类叶绿体并使其发挥光合作用的能力[6-7]。这种被吸收后且继续发挥功能的叶绿体被称为盗质体[8]。不同物种的盗质体寿命的决定因素和盗质体发挥功能的分子机制等相关的研究[9-13] 对叶绿体的内共生理论的揭示[14]以及指导植物抗逆方案的设计都具有重要的意义[15-17],但受限于光合软体动物材料的稀缺性,此相关研究的规模十分有限。白边侧足海天牛在我国的分布有利于科研人员就地取材,为我国在此方向的深入研究提供了独特的材料。分子机制的揭示离不开物种基因组的信息。海天牛属目前尚未有染色体水平上的高质量基因组的报道。基因组已经成为深入研究分子机制的关键内容。基因组含量在同一个物种里是保持稳定的[18-20]。高通量测序为一种广泛用于多种动植物基因组测序的技术[21],将高通量获得的原始数据通过质控以后,利用K-mer分析法评估基因组大小、杂合度和重复率等特征[22-23]。基因组测序大大促进了动植物的遗传进化及功能基因研究,但深度测序之前的低覆盖度的全基因组调查尤为重要,因为其可以决定基因组测序中的最合适的测序、拼装方式[24]。本实验旨在采用高通量测序技术结合K-mer分析[25],对白边侧足海天牛基因组进行测定及评估,为后续深度测序提供基础信息。
-
供试材料白边侧足海天牛(以下简称为海天牛)收集于海南省海口市东寨港红树林保护区(110°38′26″ E, 19°56′31″N),并于海南大学热带作物学院饲养,84K杨树由北京林业大学细胞生物学实验室王鑫伟提供。
-
取海天牛和84K杨树幼嫩新鲜叶片1 g,置于盛有500 μL细胞裂解液Galbraith中,并迅速将其切碎。细胞核通过30 µm滤膜后,与1 mL浓度为10 µg·mL−1的PI(碘化丙啶)染色液混合,室温孵育30 s后上机检测。使用CyFlow®Cube8(希森美康,日本)流式细胞仪对海天牛基因组大小进行评估,变异系数控制在5%以内。每个样品重复3次。
-
粉碎合格的DNA样品为350 bp左右的目的片段,构建文库,经过末端修复、加A、加接头、目标片段选择和PCR等步骤,用安捷伦2100 和定量PCR的方法检测文库片段大小和文库定量,以确定文库是否符合测序标准,通过桥式PCR的方法将文库固定到测序芯片上;将这些两端的片段在 Illumina Hiseq Xten(Illumina, U.S)测序仪上进行双末端(Paired-End)测序,获得全基因组测序数据,去除未成对匹配的读长(reads)、低质量读长、接头遭受污染以及过滤掉接头重复(duplication)的读长等对测序所产生的数据进行预处理。
-
比较原始数据与过滤后数据的质量控制数据后,使用FastQC(v 0.11.9)软件对过滤后的数据展开质量控制,包括对数据量的概览,并统计了读长每个位置测序质量,总体reads测序质量趋势,A、T、G、C碱基含量估计测序是否存在偏差,是否存在污染,数据处理时是否需要去冗余;从而实现对前期数据处理时,尽量高标准,严格质量控制。
-
通过jellyfish-2(v10.7.7)软件对序列文件进行K-mer 的计数和统计;随后,利用负二项式模型(Negative binomial model)对应的软件GenomeScope(v1.0)对基因组大小及其杂合度的评估,并生成最终基因组评估结果。选用K-mer值17、19两种条件对评估结果进行比较。
-
使用SOAPdenovo2(v2.03)软件对过滤后的数据进行拼接,拼接出Contigs序列,然后组装基因组。SOAPdenovo2的K-mer参数设置29,其他参数选择默认值。再将Contigs序列构图形成Scaffolds序列,并利用不同插入片段估计出 Contigs间的距离,用N 碱基填起来。最后,再利用测序的双末端数据之间的配对关系(Paired-End)以及短Reads数据对已组装的Contigs的覆盖信息,对Contigs间空隙(“N”)进行局部组装,补充Contigs信息,适当延长 Contigs序列。有效数据与原始序列进行对比后获得碱基深度,在序列上以5 kb为窗口,无重复前进,从而得到GC depth点图,对组装后的基因组进行评估。
-
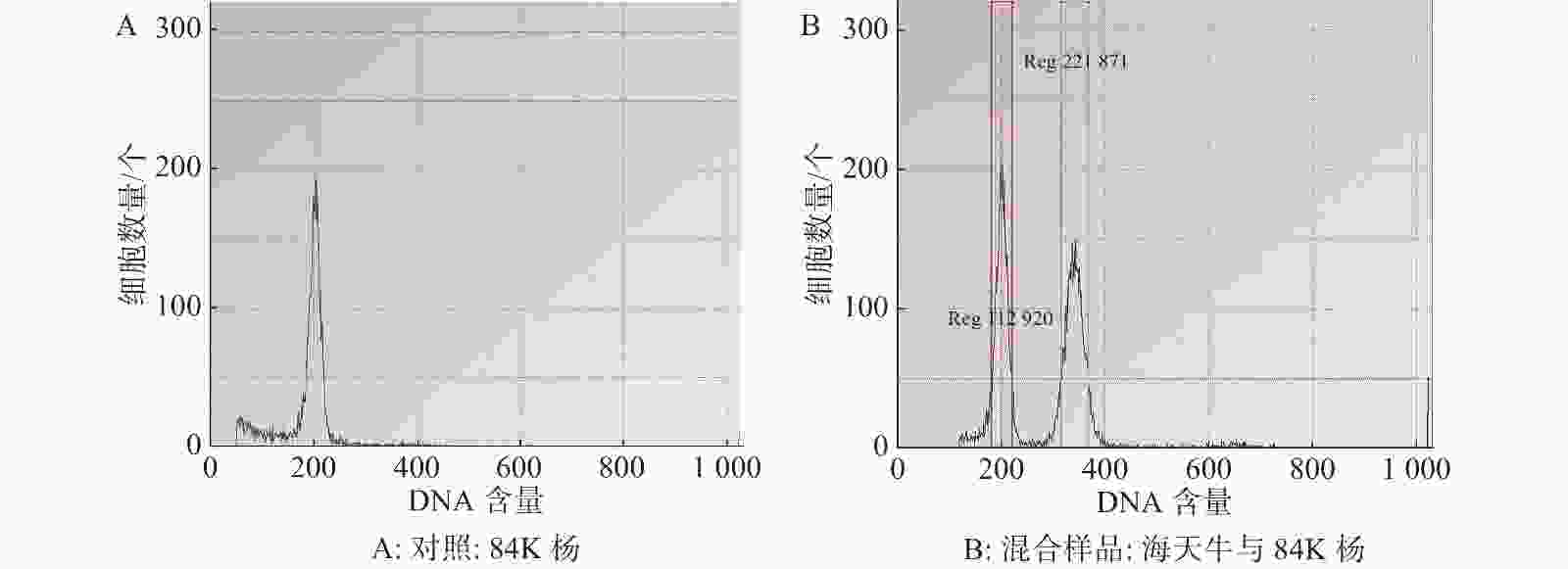
基于流式细胞术分析海天牛基因组大小,当变异系数控制在 5% 以内时,以84K杨作为对照样品(图1-A)信号峰清晰集中,84K杨与海天牛的混合样品的信号峰独立分离且距离较近(图1-B)。利用84K杨作为对照样品,根据混合样品PI 荧光强度以及峰值的倍数关系,计算海天牛基因组是84K杨的1.69倍,84K杨的核DNA相对含量为1.129 20,基因组平均值为470.155 Mb;估算出海天牛的核DNA相对含量为2.218 71,基因组平均值为794.562 Mb。
-
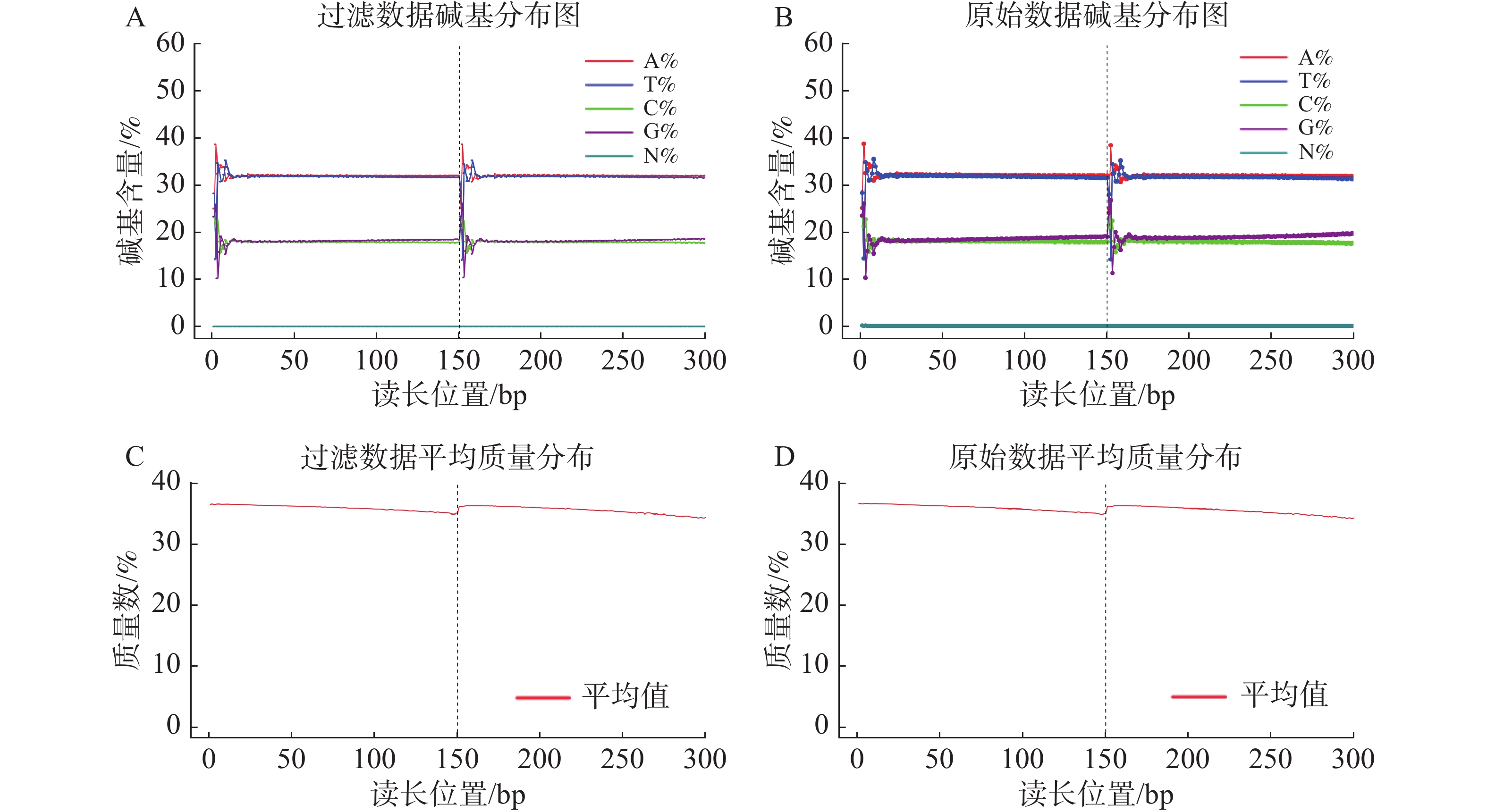
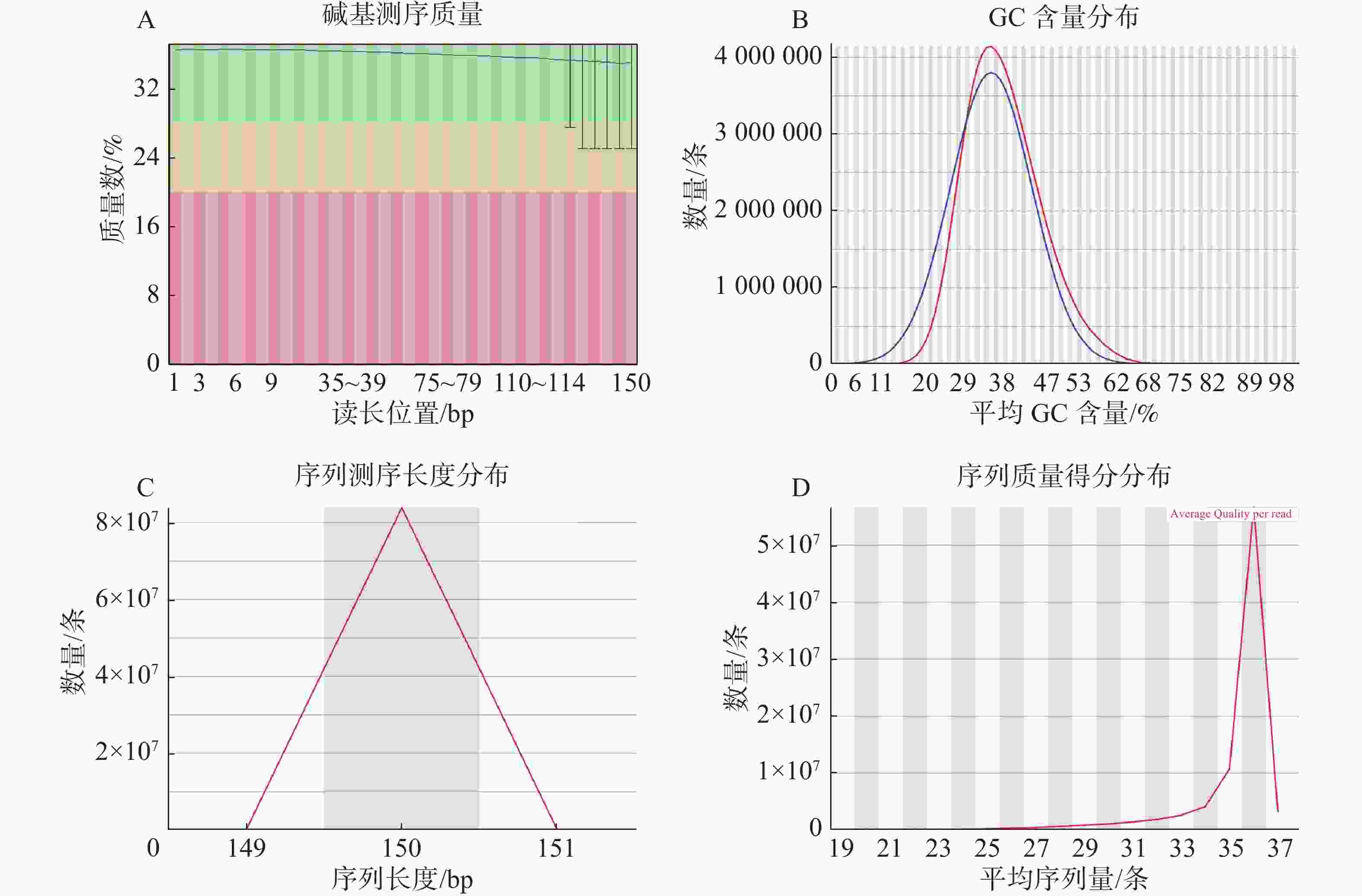
基因组调查利用第二代高通量测序技术进行双末端测序,获得全基因组的序列结果。测序共得到海天牛原始数据约25.8 Gb,共171 847 064条原始序列;过滤后约25.1 Gb,原始数据Q30比例为91.33%,过滤后Q30 比例为91.78%,满足基因组调查需要的测序数据量(表1)。比较原始数据与过滤数据(表2)的碱基的分布情况(图2-A、 2-B),过滤前后除了测序时前几个bp碱基含量略有波动属正常现象,其余每个测序位置A碱基和T碱比例相等,G碱基和C碱基比例相等,N碱基的数量为0。原始数据与过滤后数据的测序质量分布在Q30到Q40之间,Q30序列占比高,表明测序结果质量高可用于后续分析。
数据类型 数值 原始序列数/条 171 847 064 原始碱基数/bp 25 777 059 600 过滤序列数/条 167 523 764 过滤序列所占比例/% 97.48 过滤碱基数/bp 25 128 564 600 原始数据Q30比值/% 91.33 过滤数据Q30比值/% 91.78 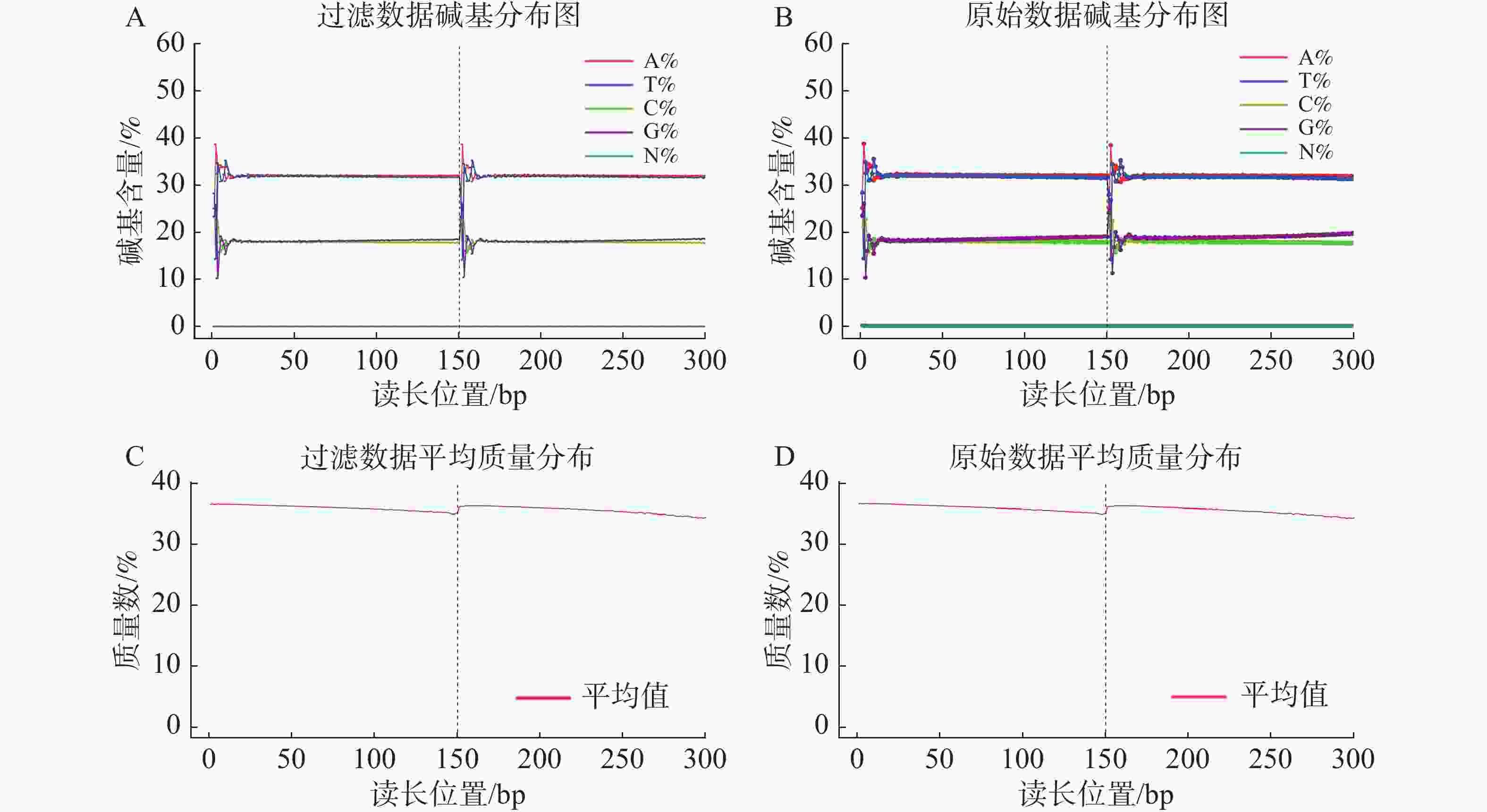
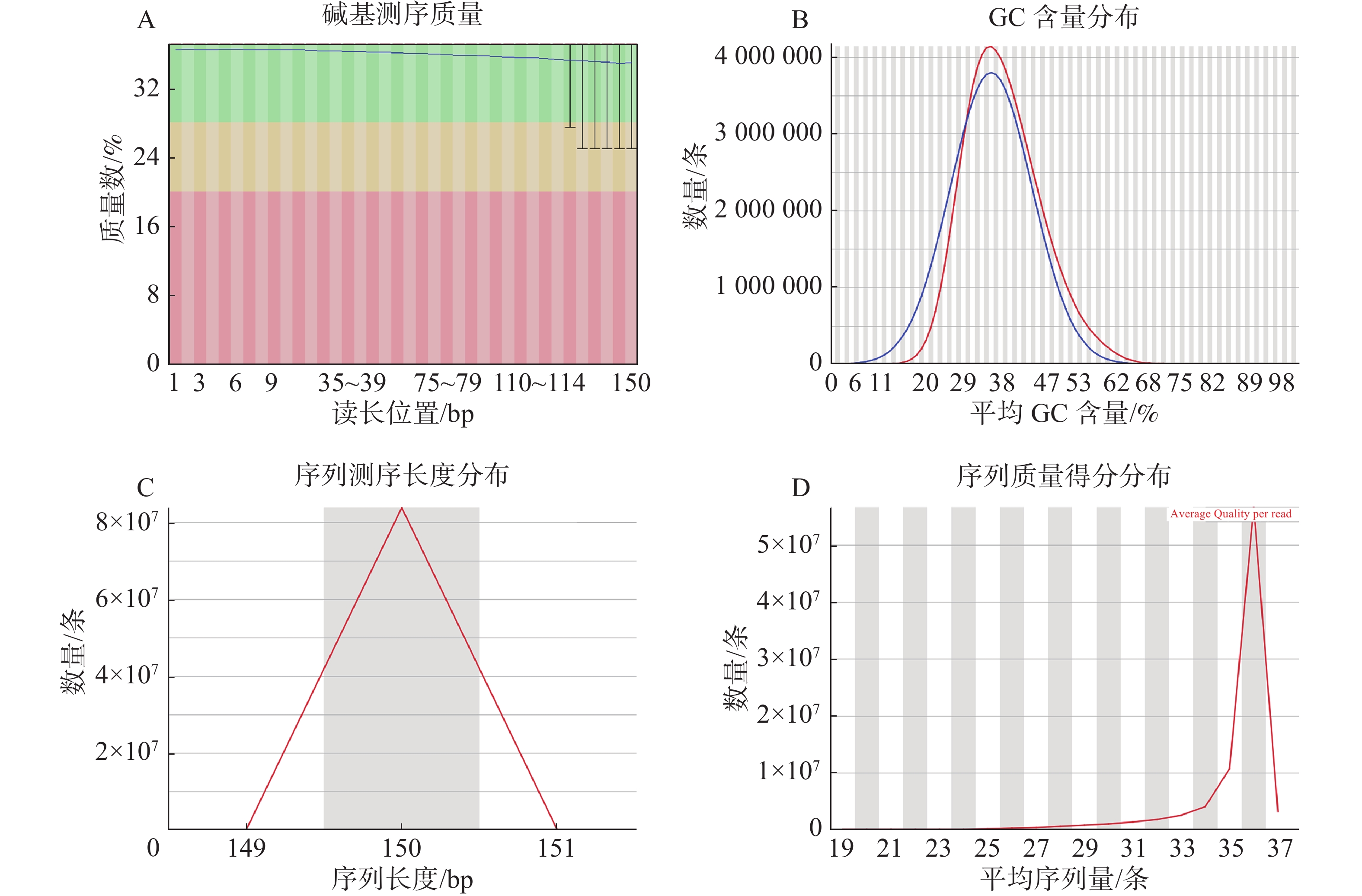
过滤数据所有读长上的碱基质量值大于30且波动小,说明过滤后数据质量稳定(图3-A);实际G、C碱基含量与理论G、C碱基含量都在38%左右出现峰值,且没有明显的偏差,表明测序结果没有偏向性(图3-B);过滤后所有的数据读长都为150 bp(图3-C);全部序列达到Q20,超过95%序列达到Q30,且集中在Q36(表2)。以上结果表明,过滤后的数据读长长,质量高,没有碱基偏好性适用于K-mer分析。
-
使用K-mer的分析方法可以预测白边侧足海天牛的基因组特征。选择K-mer的条件为17和19展开分析,样本17-mer和19-mer分布曲线为非正常泊松分布,呈现双峰分布,在17×和27×附近各有1个峰值(图3-A、图3-B)。总测序深度约为30×,根据17-mer分析,预测海天牛基因组大小约为724.8 Mb,基因组重复率为52.8%,杂合度为1.55%,模型拟合值为99.38%;19-mer分析预测海天牛基因组大小约为730.8 Mb,基因组重复率为35.1%,杂合度为1.68%,模型拟合值为99.72%(表3)。
参数 数值 总序列数/条 83761882 低质量序列数/条 0 序列长度/bp 150 G、C碱基含量/% 36 项目 17-mer 19-mer 深度 30× 30× 单倍体基因组长度/ bp 724,811,356 730,805,267 基因重复序列长度/ bp 382,653,068 256,787,169 基因特有序列长度/ bp 342,158,287 474,018,098 杂合度/ % 1.55104 1.67714 模型拟合/ % 99.3794 99.7184 读错误率/ % 0.148872 0.206886 -
选用SOAPdenovo2软件对海天牛样本进行预组装,设置K-mer参数为29时,在scaffold尺度上,得到含N碱基的基因组大小628 574 653 bp,不含N的基因组大小627 289 254 bp;Scaffold N50长度为373 bp,共405 072条;Scaffold数量2 258 693条,最长的scaffold长度为22 424 bp。在contig的尺度上,以contig N50为358 bp数量有419 361条。得到含N碱基的基因组大小624 854 764 bp,不含N的基因组大小624 854 764 bp,最长的contig为22 424 bp(表4)。组装成Scaffold的contig的数量为168 878条,每个scaffold的平均contig数目为1.5。除此,还得到scaffold尺度上的各碱基的含量,碱基A数量为207 638 986 bp,占总的碱基数目的33.03%;碱基C数量为110 706 106 bp,占总的碱基数目的17.61%;碱基G数量为109 134 885 bp,占总的碱基数目的17.36%;碱基T与碱基A的数量及占比基本相同,碱基T数量为199 809 277 bp占总的碱基数目的31.79%;剩下的所有为碱基N,数量为1 285 399 bp,占总的碱基数目的0.20%。最后计算得到G、C碱基含量为35.05%。GC-depth分析显示,测序无偏向性;平均深度集中在30×,GC 深度分布被分为2层。
类型 Scaffold
长度/bp
数量/条Contig
长度/bp
数量/条N50 373 405072 358 419361 N60 273 602827 262 624669 N70 204 870539 195 903051 N80 155 1225846 150 1271270 N90 122 1684983 120 1739030 最长片段 22424 1 22424 1 总计 628574653 2258693 624854764 2313267 -
目前,在我国已报道有8种海天牛的分布记录[1,5],其中部分海天牛具有利用藻类叶绿体进行光合作用的能力。盗质体寿命是不等的,有的盗质体能维持长达9个月[26],而有的只能维持短短的几个小时。根据叶绿体在海天牛中停留的时间将海天牛分为三类,第一类为长时间保存叶绿体物种(long-term retention (LtR) slugs),已报道的包括E. chlorotica, E. timida, E. crispata, E. clarki, E. viridis, Plakobranchus ocellatus and Costasiella ocellifera[27-30];第二类是短时间保存叶绿体物种(short-term retention species, StR),其对叶绿体的保留时间不超过两周;第三类是不保存叶绿体物种(non-retention species, NR),在食用藻类后迅速分解叶绿体[31]。在本研究中发现,白边侧足海天牛至少能保持盗质体活性2个月以上,属于能长时间保存叶绿体的海天牛。
对盗质体活性的长期保持依赖于宿主核基因编码的功能基因与盗质体基因的协调表达。例如E. chlorotica与E. timida食用藻类的叶绿体基因组中存在一种特定基因(ftsH,一种对光系统II修复至关重要的D1质控蛋白酶)其中M41金属蛋白酶结构域是维持盗质体长期活动的关键[32-33]。同时动物内源的脂肪酸合酶-(FAS)样聚酮合酶(PKS)蛋白也可以提供光保护能力,盗质体固定二氧化碳,固定碳被转化为甲基丙二酰辅酶a,并被软体动物EcPKS1酶修饰,合成紫外线-氧化阻断吡喃,保护软体动物及其叶绿体免受光合损伤[34]。另外,有观点认为吞食叶绿体后从植物中摄取的酶的丰度可能限制盗质体发挥功能。也有一种观念认为,盗质体寿命的维持是通过从藻类细胞核到动物细胞核的广泛水平基因转移(HGT)来实现。但是对于该假说还存在很大的争议,早期研究中,TORRES等证实了核编码的基因在质体核糖体抑制剂存在的条件下可以合成LHCⅠ,并提出可以通过病毒或逆转录病毒实现HTG的假说[35]。这些假说的讨论,都必须基于对海天牛核基因组及其摄取的盗质体基因组的分析研究。
E. chlorotica的全基因组测序与组装是以二代为主,三代PacBio辅助的scafford的基因组组装水平, 其全基因组大小为557 Mb,scaffold N50为442 kb,BUSCO 注释率为93.3%[36]。海天牛科目前还没有染色体水平的基因组组装结果,所以对海天牛基因组的检测仍旧是一个具有新颖性的课题。基因组调查,也称作Survey,基于深度达到20~30×以上的高质量的二代测序数据对物种的基因组大小与特征进行解读,可以为物种基因组测序方案提供重要的指导[37-38]。在基因组调查的基础上,结合流式细胞仪可提升基因组大小预测结果的精准性[39-40]。本研究中,利用84K杨为对照,通过流式细胞术预测海天牛基因组大小均值为794.562 Mb,K-mer分析的结果显示其基因组大小为724~730 Mb,两者的结果偏差较小,不影响对基因组测序决策的判断 [41]。所有的结果显示,白边侧足海天牛是一个高度杂合的物种,且基因组大小超过700 Mb。为了达到染色体级别的组装水平,全基因组测序建议使用以三代测序技术为主,Hi-C或Hi-Fi技术相结合的测序手段[42-44],测序量达到80×~100×的深度足够完成海天牛基因组的精细组装。
Whole genome survey analysis of Elysia leucolegnote
DOI: 10.15886/j.cnki.rdswxb.2022.05.005
- Received Date: 2021-11-04
- Accepted Date: 2022-03-31
- Rev Recd Date: 2022-04-12
- Available Online: 2022-07-12
- Publish Date: 2022-09-21
-
Key words:
- Elysia leucolegnote /
- genomic survey analysis /
- genome size /
- heterozygosity
Abstract: Elysia leucolegnote are sacoglossan sea slugs, belonging to the family Elysiidae and the genus Elysia. They are distributed in mangroves throughout China, Thailand and the Philippines. Photosynthetic sea slugs are famous for their ability of photosynthesis. Stolen chloroplasts are called kleptoplasts. However, there has been no important breakthrough in the molecular mechanism research of kleptoplasts. In this context whole genome survey analysis of E. leucolegnote was conducted to find the clue about the regulation of photosynthesis and maintenance of chloroplast activities by high-throughput sequencing technology. The high-throughput sequencing produced 25 Gb high-quality data in E. leucolegnote, with a total sequencing depth of about 30x. According to 17-mer analysis, the predicted genome size was 724.8 Mb, the genome duplication rate was 52.8%, the heterozygosity was 1.55%, and the model fitting value was 99.38%. The 19-mer analysis predicted that the genome size was 730.8 Mb, with the genome duplication rate of 35.1%, the heterozygosity of 1.68%, and the model fitting value of 99.72%. A draft genome of E. leucolegnote Ker was obtained by preliminary assembly with a total length of 628 Mb. These results showed that E. leucolegnote is a highly heterozygous species with a genome size of more than 700 Mb.
| Citation: | LI Xinyu, DING Mengying, FENG Erhui, ZHANG Yuanyuan, ZHANG Kelan, WAN Yinglang. Whole genome survey analysis of Elysia leucolegnote[J]. Journal of Tropical Biology, 2022, 13(5): 457-463. doi: 10.15886/j.cnki.rdswxb.2022.05.005

|
 DownLoad:
DownLoad: