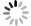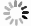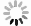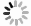

-
丙氨酸脱氢酶(alanine dehydrogenase,ALD,EC 1.4.1.1)是一种氧化还原酶,使丙氨酸可逆转化为丙酮酸[1]。编码ALD的基因为ald基因。结核分枝杆菌(M.tuberculosis)的ald基因在E. coli中表达后具有丙酮酸胺化活性;而当ald基因在M. tuberculosis中失活时,会导致细菌ALD活性丧失[2]。因此,ald基因是微生物中ALD活性必需的[1]。丙氨酸脱氢酶在自然界中分布广泛,存在于细菌、植物和真核藻类中,在微生物的氮、碳代谢中起重要作用[3]。ALD将脱氨/胺化作用与核苷酸辅因子(NAD+)的还原/氧化联系起来,从而与微生物中多种能量消耗/产生过程相关[4]。丙氨酸脱氢酶在孢子形成过程中以L−丙氨酸为主要能源,通过柠檬酸循环供应能量[5],同时参与固氮作用(氨同化),确保生物体内氮源充足[6-7],也可以平衡机体内的糖代谢和氨基酸代谢[8]。此外,丙氨酸脱氢酶作为新的抑制剂靶标也具有特殊的医学意义[9],抑制ALD会导致肽聚糖层合成所需的L−丙氨酸不足,从而阻碍致病细菌的生长[10]。因此,基于丙氨酸脱氢酶的生物学特性开发的生物抑菌剂,能够对细菌尤其是致病菌的生长产生一定的抑制作用[11]。
维氏气单胞菌(Aeromonas veronii)是一种革兰氏阴性菌,广泛存在于自然环境中,是感染鱼类的最常见致病菌之一。目前已证实,维氏气单胞菌可感染罗非鱼、斑点叉尾、草鱼等20多种水生生物,病鱼主要有脏器出血、严重腹水等症状[12]。此外,维氏气单胞菌还能够感染狐、熊猫等哺乳动物,导致其精神沉郁,肠胃炎,肝脏病变[13]。该菌也可以导致人类疾病,人食用了被污染的水产品和畜禽产品后,可引发腹泻、脑膜炎和败血症等,免疫力低下者甚至死亡[14]。因此,维氏气单胞菌被定义为一种人−兽−鱼共患的致病菌,从而引起人们的警惕与重视。为了后续在维氏气单胞菌中开展ALD相关研究,开发潜在特异性抑菌剂,笔者通过基因重组技术成功构建Flag-ALD融合表达质粒,在大肠杆菌表达体系中对其进行诱导表达,并通过SDS-PAGE和western blot技术检测外源表达蛋白。本研究构建的重组载体为后续进一步探究ALD蛋白在维氏气单胞菌中的调控功能夯实了基础,有利于揭示丙氨酸脱氢酶在各种代谢调控通路中起的关键作用。以ALD为新的切入点,有助于揭示维氏气单胞菌的致病机制,为预防和治疗由该病原菌引起的水产病害提供新的途径和思路。
HTML
-
本实验用的菌株与质粒见表1。
实验材料 Burst material 相关属性 Related attributes 来源 Source 菌株
StrainsE. coli BL21 野生型,用于表达外源蛋白表达 实验室保存 E. coli DH5α 野生型,用于质粒克隆 实验室保存 Aeromonas veronii 野生型,分离自发病鱼体 实验室保存 质粒
PlasmidspACYDuet 大肠杆菌蛋白表达载体,含有左霉素抗性基因 实验室保存 pACYDuet-Flag-ald pACYDuet重组质粒,包含T7启动子和维氏气单胞菌融合Flag标签的 ald 基因 本实验构建 Table 1. Strains and plasmids used in this study
-
限制性核酸内切酶NcoⅠ,EcoRⅠ和T4DNA连接酶选购自美国NEB公司;2×PhantaMaxMaster Mix,DL5000 DNA Marke、质粒提取试剂盒、胶回收试剂盒、PCR纯化试剂盒购自中国Vazyme公司;双色预染蛋白Mark、5×蛋白质加样缓冲液购自中国生工公司;用于Western blot的anti-Flag抗体、山羊抗小鼠lgG H&L(Alexa Fluor ®488)购自美国abcam公司;其他试剂均为国产分析纯。
-
把维氏气单胞菌C4的基因组DNA作为模板,用引物对Flag-ALD-F/ALD-R扩增ald目的基因(Flag-ALD-F:5′-CATGCCATGGGCGATTACAAGGATGACGACGATAAGATGATTATCGGTGTACCTAA-3′;ALD-R:5′-CGGAATTCTCAGTTCAGCAGGGTCAGGG-3′)。PCR反应体系为50 μL,体系包括:上下游引物各1 μL,模板1 μL,2×PCR mix25 μL,ddH2O 22 μL。PCR扩增条件为:预变性94 ℃ 10 min,变性94 ℃ 30 s,退火55 ℃ 30 s,延伸72 ℃ 80 s,总计循环30次,终延伸72 ℃ 10 min,4 ℃停止反应。PCR产物用1%琼脂糖凝胶电泳分离检测,对目的条带进行割胶纯化回收。
-
将纯化后的目的片段与pACYCDuet空载质粒分别用限制性核酸内切酶NcoⅠ与EcoRⅠ进行双酶切处理,酶切体系为50 μL,包括:内切酶NcoⅠ和EcoRⅠ各1 μL,Cutsmart缓冲液5 μL,目的基因和质粒各600 ng,ddH2O 30 μL。将酶切产物用试剂盒纯化回收并用T4 DNA连接酶放在22 ℃金属浴中连接30 min,连接体系包括:pACYCDuet质粒30 ng,目的基因90 ng,T4 DNA连接酶1 μL,Cutsmart缓冲液1 μL,ddH2O 10 μL。将连接产物电转化至已经制备好的E. coli DH5α感受态细胞内,将转化细菌在37 ℃复苏1 h后涂布到含25 mg·L−1左霉素的LB固体培养基上,37 ℃恒温培养过夜后,挑取转化子的单菌落至LB培养基中振荡培养,提取质粒。用载体特异性引物对pACYCDuet-F/pACYCDuet-R进行菌落PCR(pACYCDuet-F:5′-TAATACGACTCACTATAGGG-3′;pACYCDuet-R:5′-TGCTAGTTATTGCTCAGCGG-3′),以验证阳性转化子,将验证正确的阳性转化子送至上海生工生物工程有限公司测序。
-
将测序成功的pACYCDuet-Flag-ald重组载体电转化至E. coli BL21感受态中,挑取单菌落至含25 mg·L−1 氯霉素的5 mL LB培养基中37 ℃恒温振荡培养12 h。将其菌液按初始OD600值为0.01的接种量转接到200 mL LB培养基中(含25 mg·L−1氯霉素),37 ℃培养至OD600值为0.4时加入终浓度0.1 mmol·L−1的异丙基−β−D−1−硫代半乳糖吡喃糖苷(IPTG)诱导外源蛋白大量表达,每隔1 h取菌液,6 000 r·min−1离心10 min收集菌体,在收集的菌体中加入50 μL 1×蛋白上样缓冲液(250 mmol·L−1 Tris-HCl,pH 6.8,10%SDS,0.5%溴酚蓝,50%甘油,7.5%DTT),吹打混匀后,100 ℃金属浴20 min,13 000 r·min−1离心10 min,分离上清和沉淀。向沉淀中加入20 μL 1×蛋白加样缓冲液,吹打混匀后,100 ℃水浴锅20 min。各取10 μL上清和沉淀蛋白提取液,用12%SDS-PAGE凝胶进行电泳分析,考马斯亮蓝R-250染色1 h显示电泳结果。
-
将含有外源表达蛋白的细胞全蛋白提取物通过12%SDS-PAGE凝胶进行电泳分离,在250 mA的恒定电流下通过湿转法转膜90 min,将全蛋白提取物转移到NC膜上。用5%脱脂奶粉封闭液(10 mmol·L−1 Tris-HCI,100 mmol·L−1 NaCl,25 mmol·L−1 NaF,0.1%Tween-20,pH7.4)室温封闭硝酸纤维素膜3 h,然后用anti-Flag抗体4 ℃孵育过夜,TBST (10 mmol·L−1 Tris-HCI,135 mmol·L−1 NaCl,2.5 mmol·L−1 KCI,0.1%Tween-20,pH 7.4)洗膜3次,每次10 min,之后用荧光标记的山羊抗小鼠lgG H&L(Alexa Fluor ®488)避光常温孵育2 h,1X TBST洗膜3次,每次10 min,最后在Typhoon FLA 9500(GE,USA)多功能激光扫描仪上扫描荧光标记的NC膜,检测目的蛋白表达情况。
1.1. 实验材料
1.2. 实验试剂
1.3. Flag-ald 目的基因的扩增及纯化
1.4. pACYCDuet-Flag-ald 重组载体构建
1.5. Flag-ALD融合蛋白的诱导表达及Western blot分析
1.5.1. Flag-ALD融合蛋白的诱导表达及SDS-PAGE凝胶电泳分析
1.5.2. Western blot分析
-
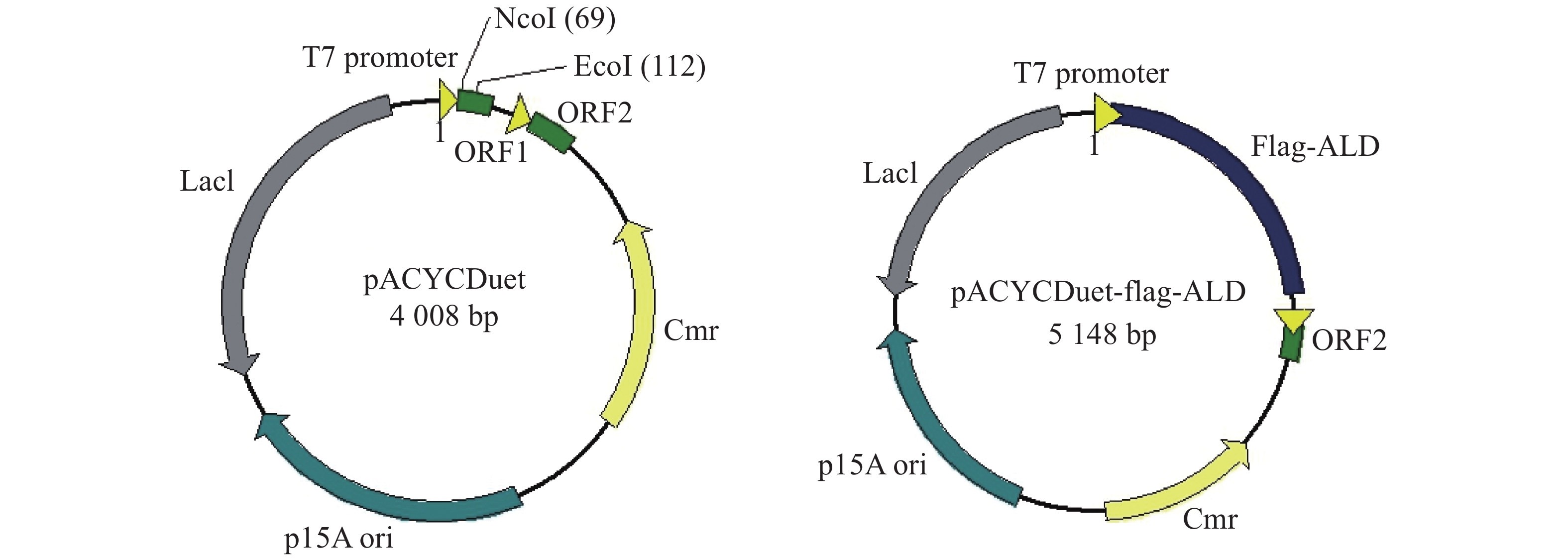
为了便于检测在E. coli中大量表达的ALD蛋白,笔者在ald基因N端融合Flag标签,通过检测Flag标签来验证ALD蛋白的表达。E. coli BL21是常用的大肠杆菌工程菌株,通常与含有T7启动子的质粒搭档,用于诱导蛋白的过量表达。从图1可知,本实验选择pACYCDuet载体在E. coli BL21中表达Flag-ALD蛋白,质粒含有T7启动子,经IPTG诱导可大量表达目标蛋白。
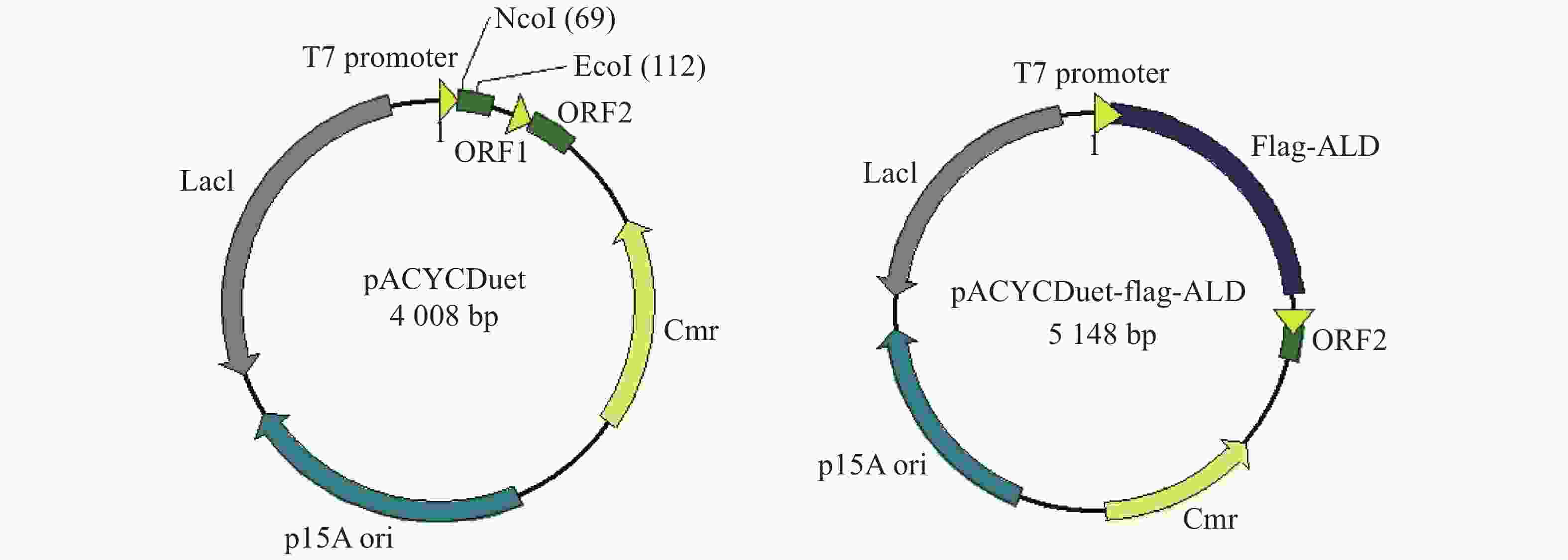
Figure 1. The plasmid profiles of pACYCDuet and pACYCDuet-Flag-ALD
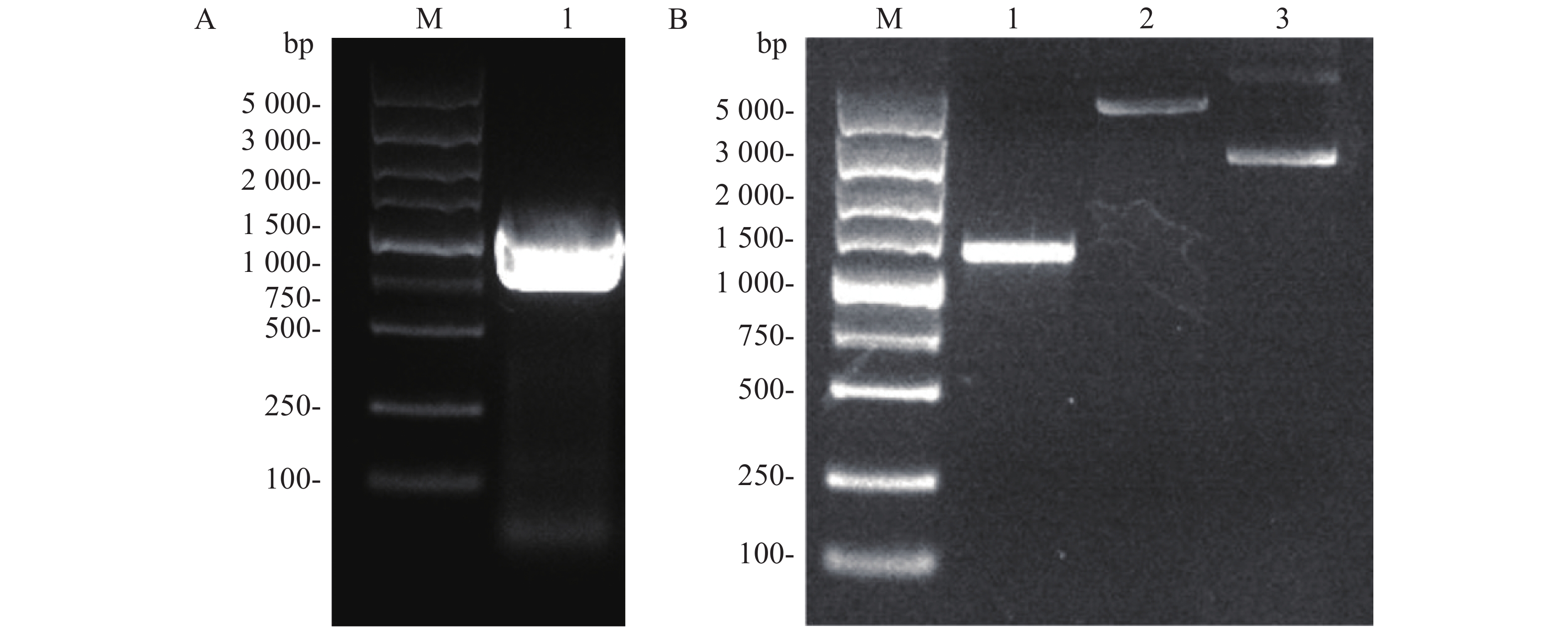
Flag标签含有8个氨基酸,核苷酸序列为24 bp,将其核苷酸序列并入用于扩增ald基因的上游引物的5'端,经PCR扩增即可获得Flag-ALD融合片段。ald基因序列长度为1 116 bp,则编码Flag-ALD融合片段的核苷酸序列理论长度为1 140 bp。PCR产物经过1%琼脂糖凝胶电泳分析,结果显示,大约1 100 bp处存在1条明亮单一的目的条带,可用于后续实验(图2A)。通过割胶回收纯化PCR产物后,与pACYCDuet空载体均用限制性核酸内切酶NcoⅠ和EcoRⅠ进行双酶切,并对酶切产物进行纯化回收。对酶切产物进行1%琼脂糖凝胶电泳分析(图2B),从图2B可知,酶切后的目的片段长度在1 000~1 500 bp之间(泳道1),酶切后载体的泳动速度比酶切之前载体的泳动速度慢(泳道2和泳道3),表明载体已被成功酶切,可用于后续实验。
Figure 2. PCR amplification of ALD gene (A) and double restriction digestion of the target gene and pACYCDuet plasmid (B)
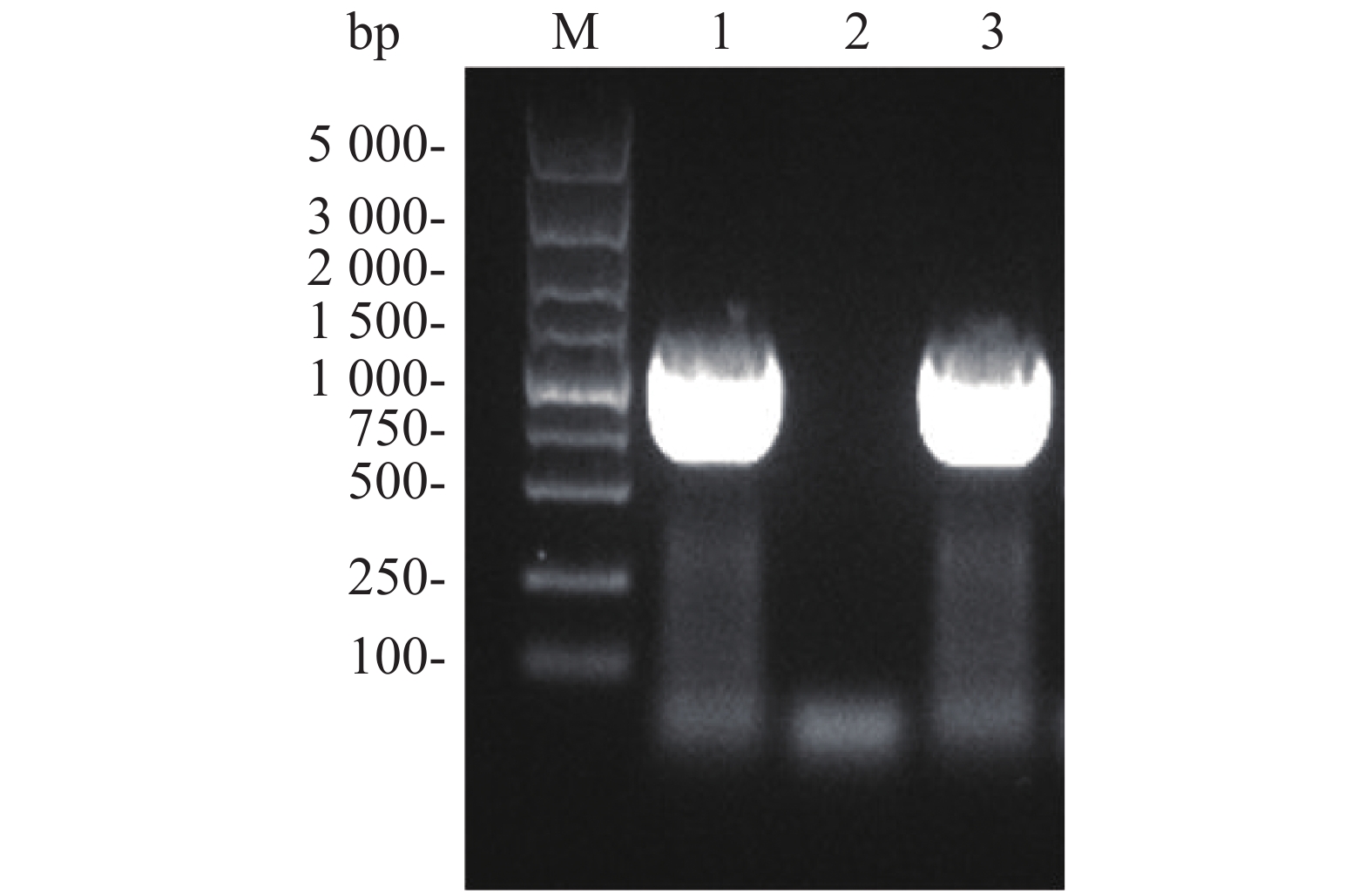
通过酶切连接构建融合表达载体pACYCDuet-Flag-ald,将其电转化至E. coli DH5α 感受态中,并通过含25 mg·L−1 左霉素的抗性平板筛选阳性克隆。挑取候选阳性克隆菌落,以其为模板进行菌液PCR,以验证阳性转化子。菌液PCR结果表明,以阳性克隆载体为模板,能够扩增获得大约1 000 bp的目的产物(图3,泳道1和3)。从阳性转化子中提取质粒,由上海生工生物工程有限公司测序,测序结果表明,重组载体构建成功。
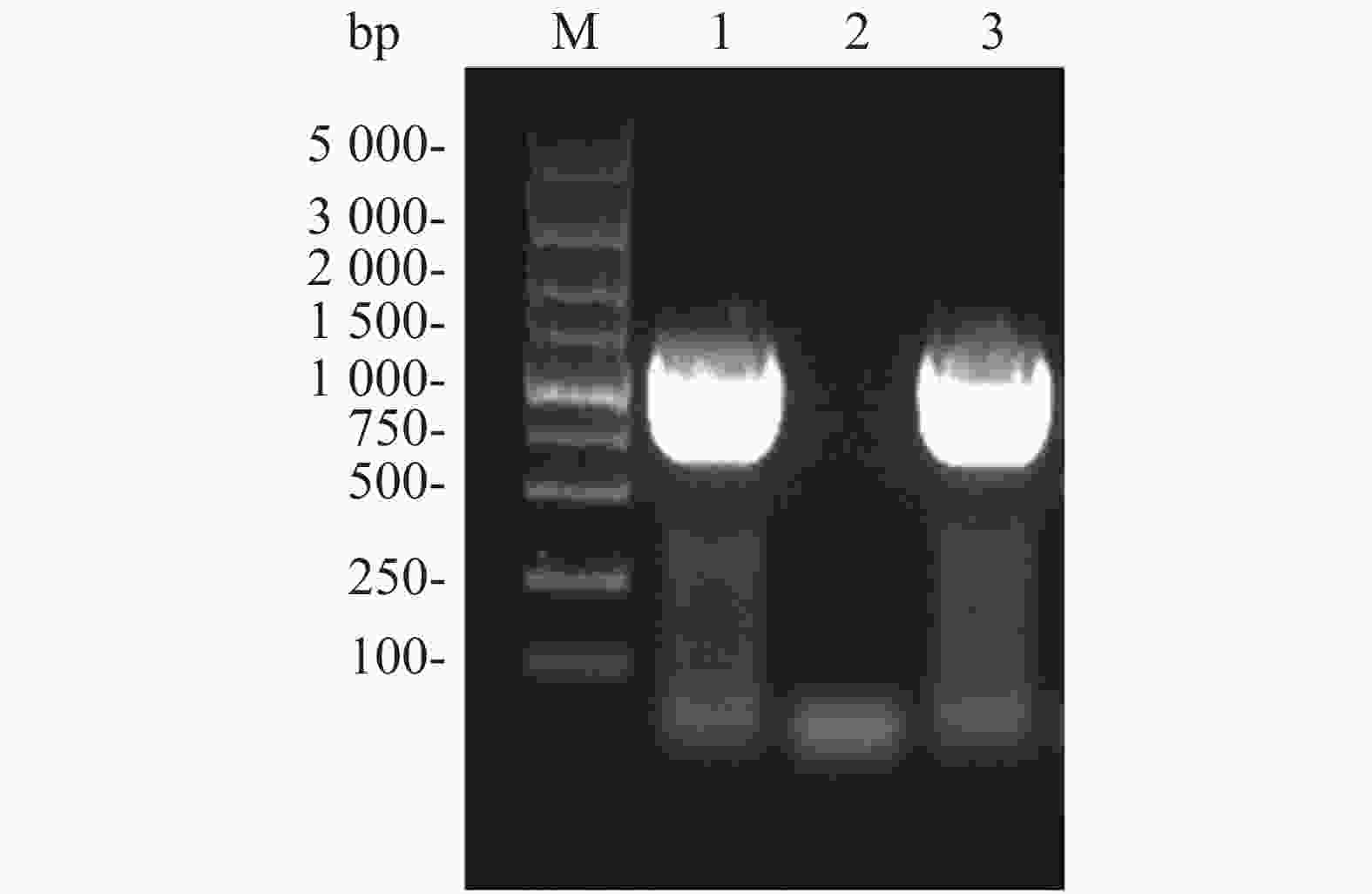
Figure 3. Identification of positive clones by bacterial PCR
-
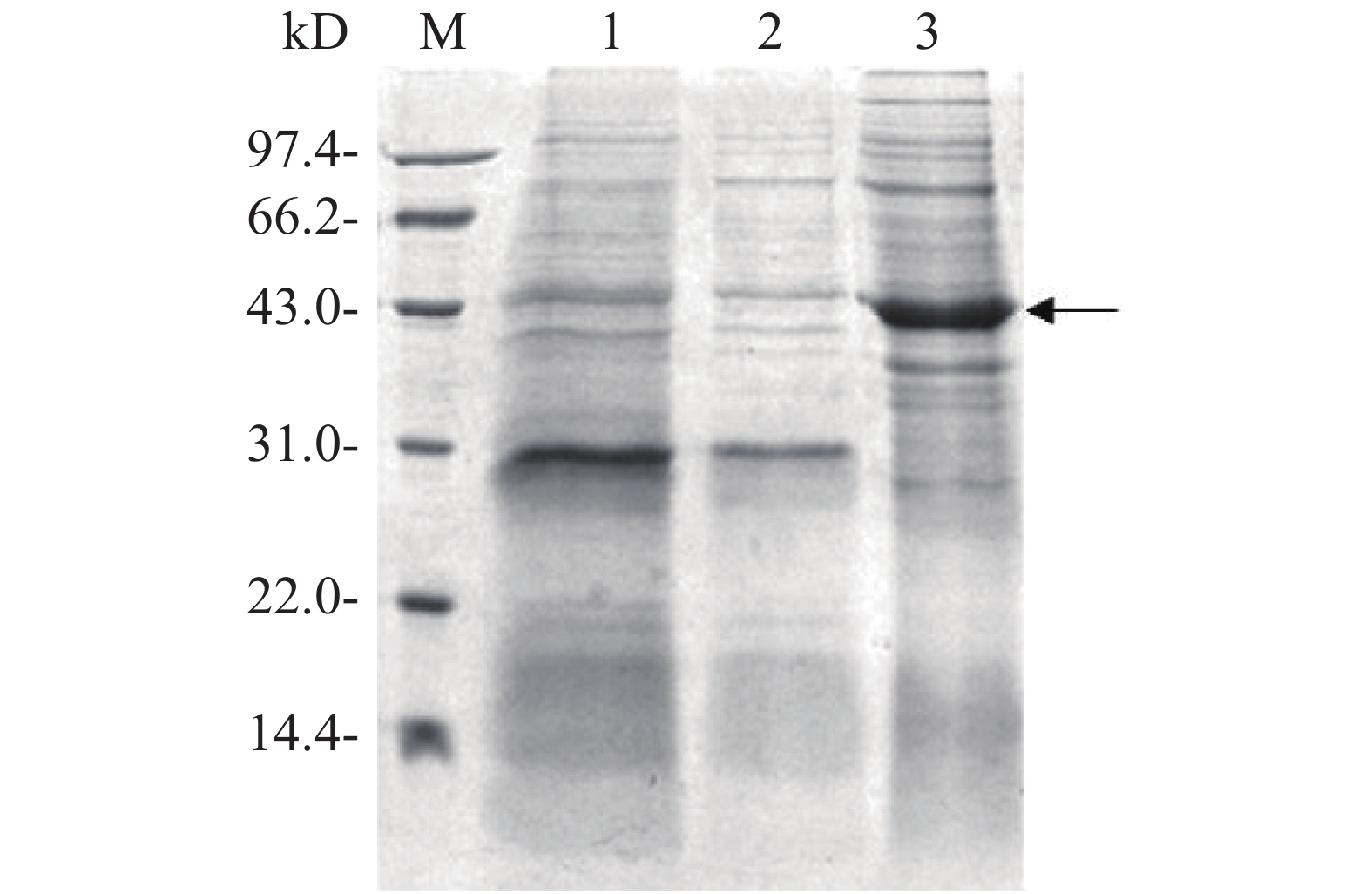
将测序结果正确的融合表达载体电转化至E. coli BL21中,加入0.1 mmol·L−1 IPTG诱导目的蛋白表达。用热裂解法裂解细菌,超速离心后,分别收集上清液和沉淀进行SDS-PAGE凝胶电泳分析,对凝胶进行考马斯亮蓝染色后观察,从图4可知,与没有添加IPTG诱导组(泳道1)相比,加入IPTG诱导2 h后,菌体裂解上清液在位于约42 kD处(泳道3)出现显著的目的蛋白条带(图4箭头所示),而沉淀中没有观察到目的蛋白表达(泳道2),表明融合蛋白Flag-ALD以可溶性形式成功表达。
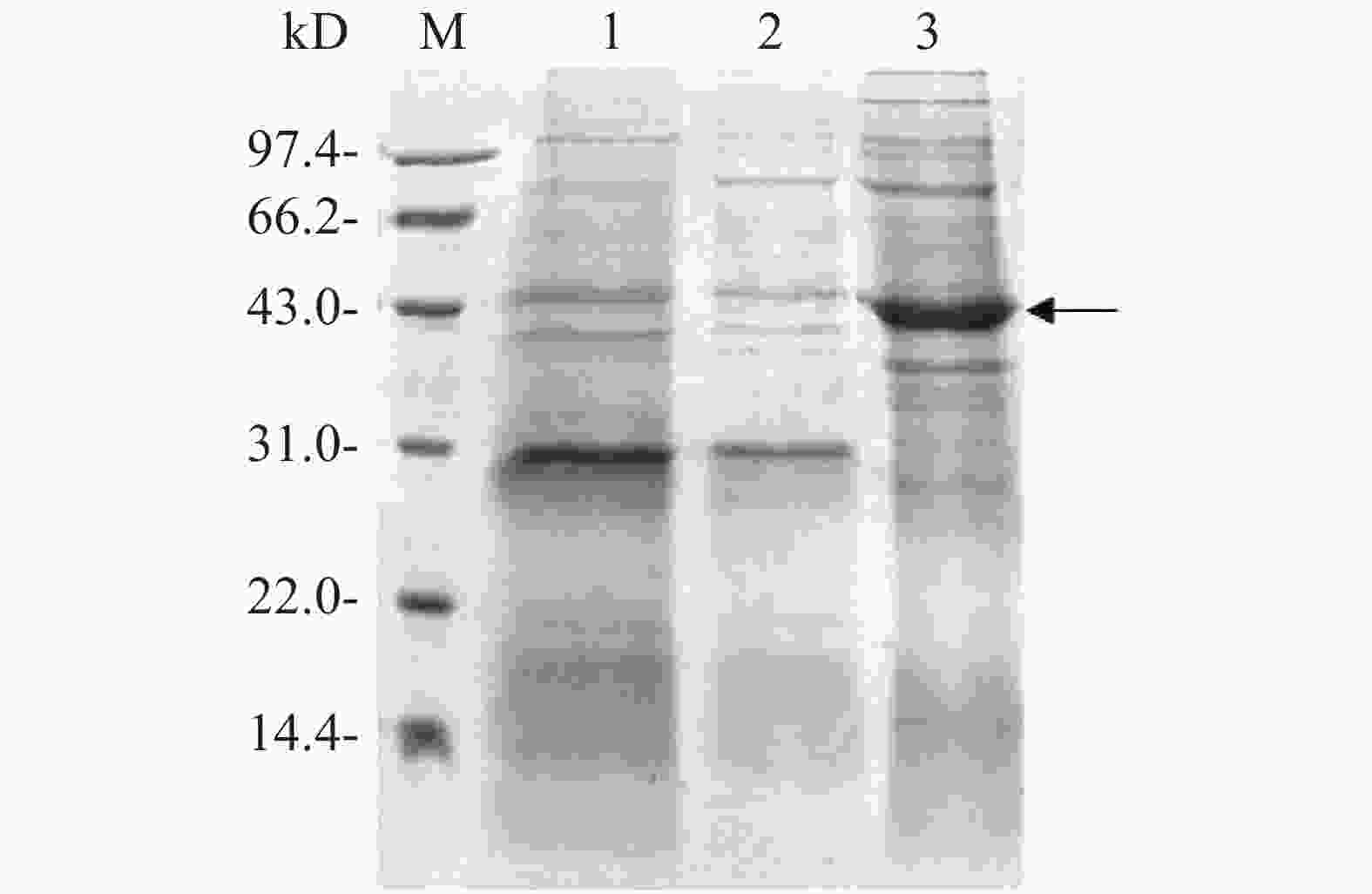
Figure 4. Induced expression of the fusion protein Flag-ALD
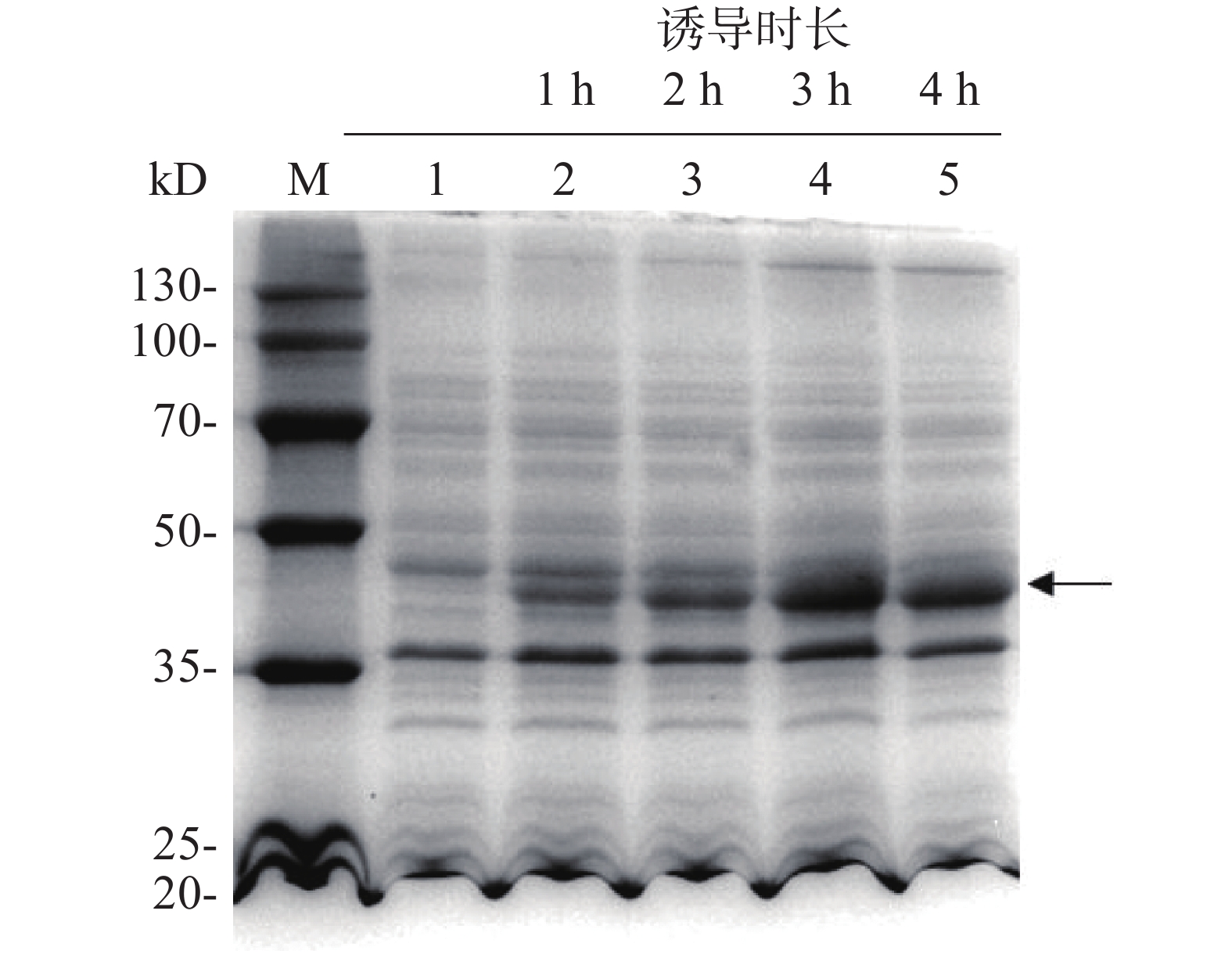
为了探索IPTG诱导的最佳时间,每隔1 h取1次菌液,用热裂解法裂解细菌,离心后取上清液进行SDS-PAGE凝胶电泳分析,随后考马斯亮蓝染色,结果表明(图5),与未添加IPTG的组(泳道1)相比,加入IPTG诱导后(泳道2~5),伴随着诱导时间增加,诱导表达的目的蛋白浓度逐渐增加(箭头所示),且诱导3 h后即使诱导时长增加,蛋白表达量也保持大致相同。
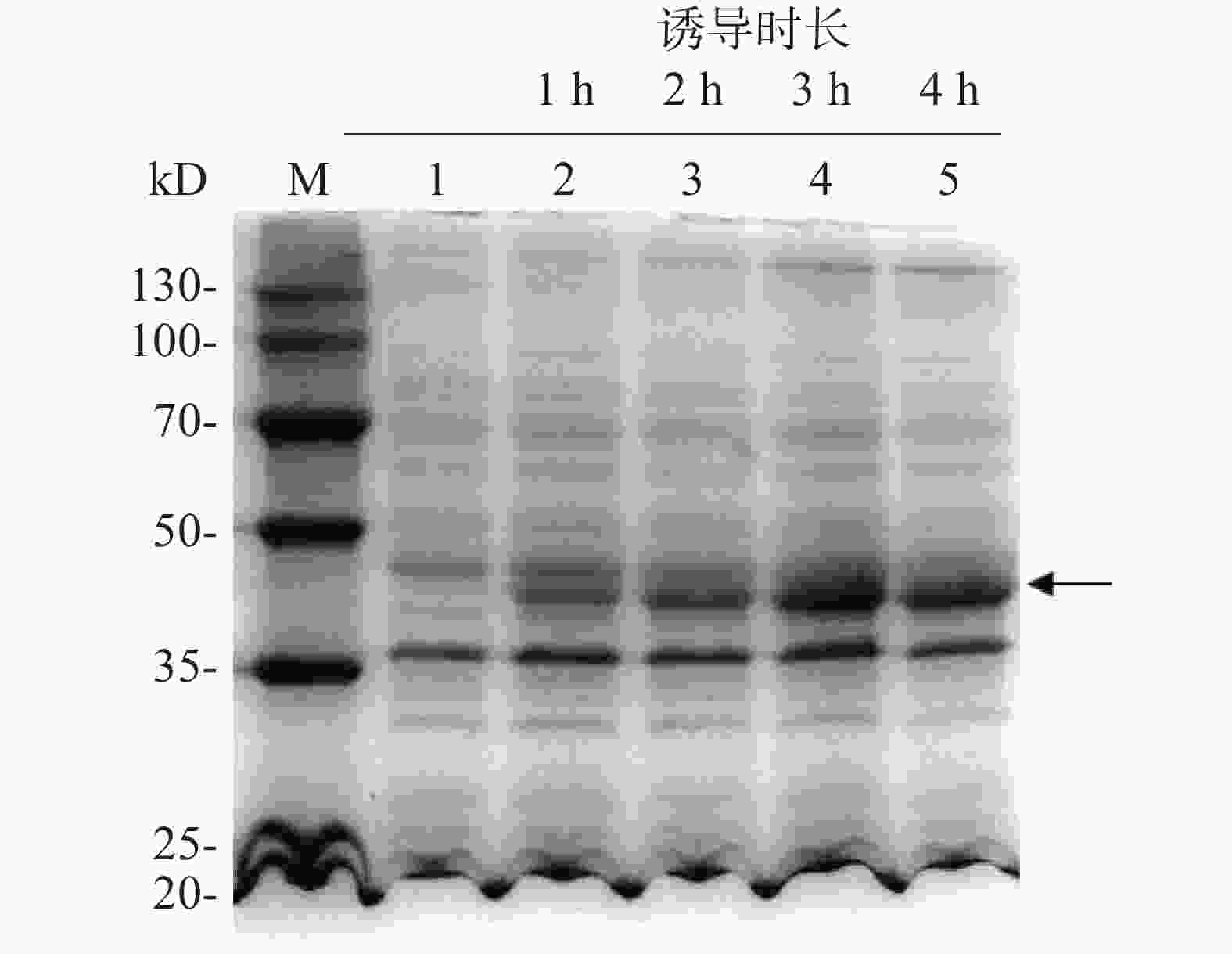
Figure 5. Effect of IPTG-based inducing time on the expression of target protein
-
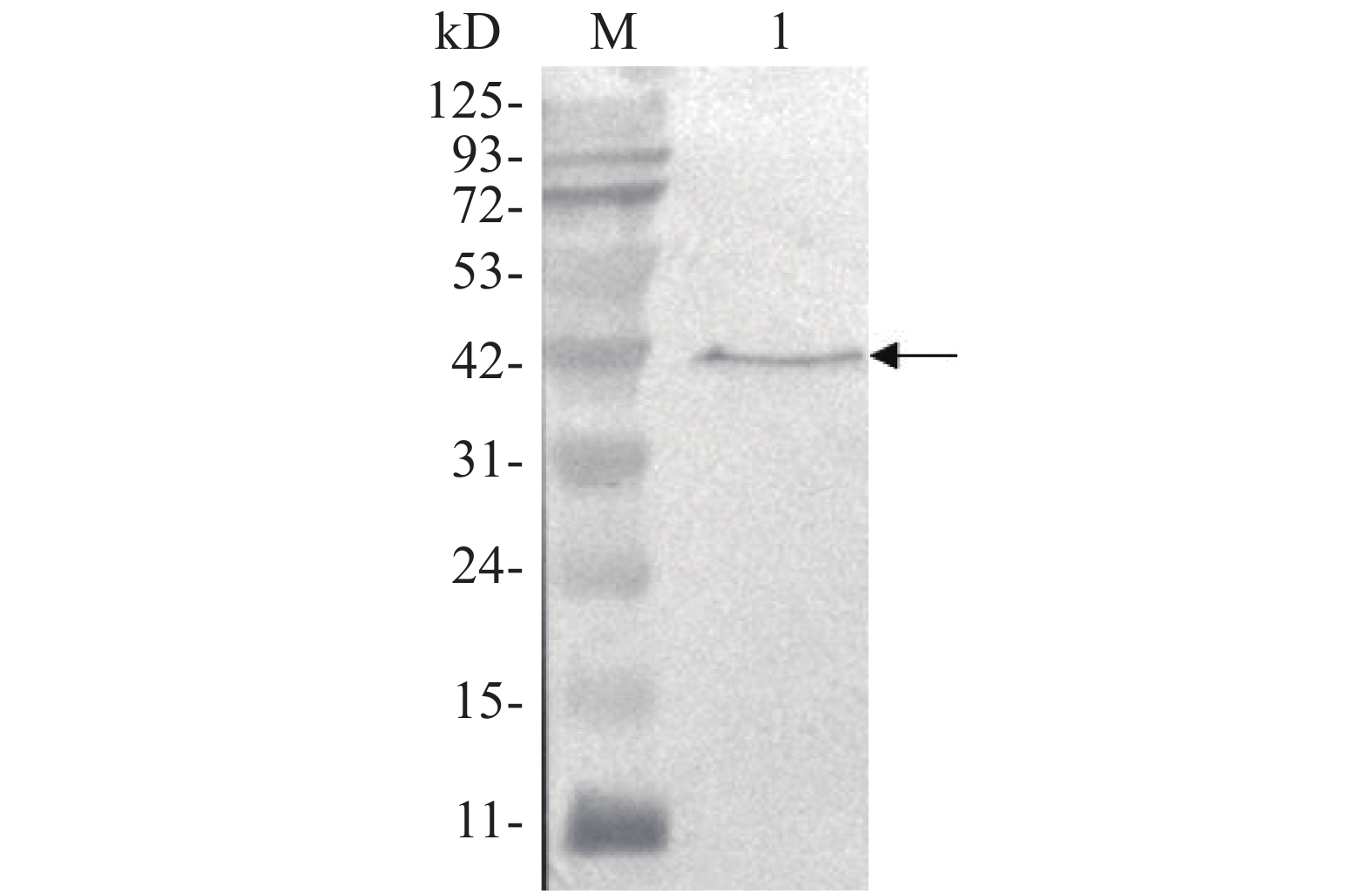
用anti-Flag抗体对携带Flag标签的靶标蛋白进行Western blot,结果在42 kD处检测到1条特异条带(图6),Flag-ALD融合蛋白的相对分子质量为41.8 kD,Western blot结果与预期相符,说明Flag-ALD目的蛋白已经成功表达。
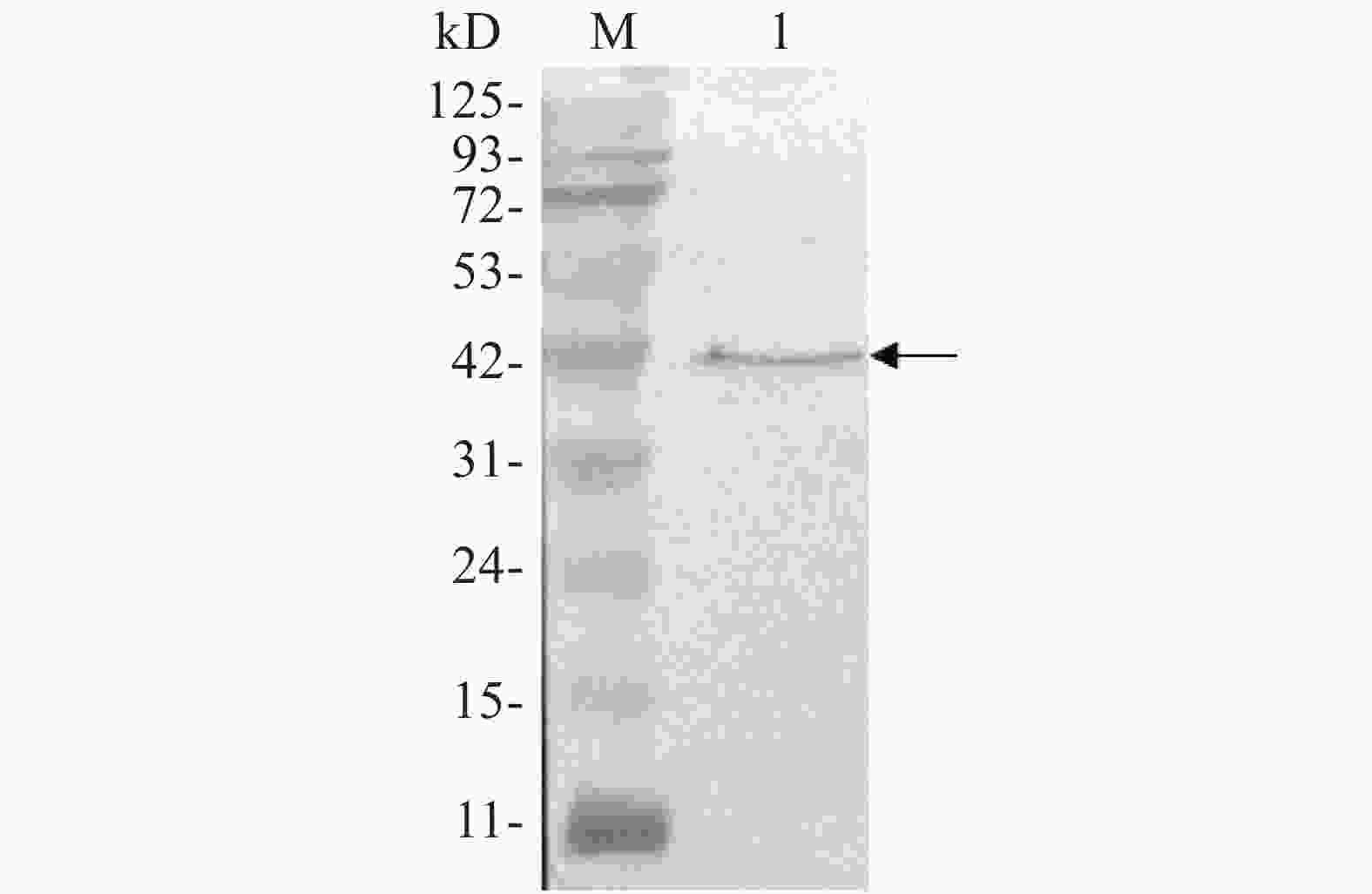
Figure 6. Detection of the expression of Flag-ALD fusion protein by Western blot
2.1. pACYCDuet-Flag-ald 表达载体构建及鉴定
2.2. 融合蛋白Flag-ALD诱导表达
2.3. Western blot鉴定Flag-ALD蛋白的表达
-
维氏气单胞菌是一种新型的人、鱼共患的病原菌,致病性强,可导致广泛的水生生物感染,对水源生物和人类健康存在一定的危害[13]。因此,研究维氏气单胞菌的致病机制,寻找抑制剂靶标,研究新型抑菌剂变得尤为重要。丙氨酸脱氢酶是一种生理酶,ALD在微生物体内的主要作用为产生丙氨酸[15],为肽聚糖的合成提供原料[16]。研究表明,巨大芽孢杆菌[17]和嗜碱性假坚强芽孢杆菌[18] OF4中的ALD蛋白都曾在大肠杆菌中外源表达,通过基因克隆技术构建原核表达载体pET-OF4Ald,经硫酸铵分级沉淀、分子筛柱层析等方法获得纯化后蛋白。上述研究为维氏气单胞菌来源的ALD蛋白在大肠杆菌中外源表达提供了理论基础,但均未对外源蛋白进行标记,需要使用较为复杂的纯化方法,而且也不利于后续直接检测外源蛋白的表达
本实验以维氏气单胞菌的丙氨酸脱氢酶为研究对象,通过分子克隆获得Flag-ALD融合表达载体,并在大肠杆菌中大量表达该蛋白。由于ALD的单克隆抗体没有商品化且制备周期较长,为了方便ALD蛋白的检测,本研究采取的策略是在其N端融合Flag标签,通过检测Flag标签就可以反映ALD蛋白在大肠杆菌中表达的表达情况。选择Flag标签作为融合标签是因为Flag标签全长8个氨基酸,分子质量不大,对目的蛋白的空间结构和表达没有影响,且anti-Flag抗体较常见,易获得。通过SDS-PAGE凝胶电泳分析可知,得到的重组蛋白ALD以可溶性蛋白的形式存在,菌体裂解后的沉淀中基本不含目的蛋白。通过对诱导蛋白表达条件的改良可知,在加入0.1 mmol·L−1 IPTG诱导3 h后,目的蛋白的表达量基本达到最大,这为后续实验需大量表达目的蛋白提供了最佳表达条件。本研究获得的ALD外源表达蛋白,可用于开发生物抑菌剂,以酶蛋白为靶标,开发合适的抑菌剂从而有效地抑制细菌滋生,为开发病原菌维氏气单胞菌的生物抑制剂提供了新的切入点。同时,也为病原菌的致病机制及病害防治提供了一条研究途径。
 DownLoad:
DownLoad: