
-
昆虫是自然界物种较丰富、数量庞大的生物类群[1],已知种类超百万种[2]。蚤类是重要的医学昆虫,为专性吸血寄生虫,在全球广广泛分布;蚤的叮咬会引发宿主过敏性皮炎、贫血,干扰人和动物的正常生活,更对兽医公共卫生构成显著威胁[3]。但蚤种的准确鉴定是实践中的一大挑战。传统形态学鉴定对形态相似种、受损标本、幼虫及属内雌性标本等的鉴定难度极大[4-5]。DNA条形码技术以COI基因为核心分子标记,可实现蚤种的快速、准确鉴定,已成为形态学分类不可或缺的补充与验证工具[6]。鉴定技术的进步推动了蚤类体内微生物的发掘,目前已在蚤体内检出鼠疫耶尔森菌(Yersinia pestis)、立克次体属(Rickettsia)[7]及巴尔通体属(Bartonella)[8]等多种病原微生物。高通量测序(High-throughput sequencing HTS),(Next-Generation Sequencing NGS)[9]作为变革性生物技术,可实现DNA分子的大规模平行测序,且无需预设序列信息,能在复杂基因背景中精准识别病原序列,现已广泛应用于各类生物检测领域[10],也为全面解析蚤类微生物群落特征提供了关键技术支撑。
海南岛位于北纬18°10'~20°10',东经108°37'~111°03'之间[11],属热带海洋性季风气候,常年高温高湿的环境为蚤类繁殖提供了有利条件,大幅提升了当地蚤传疾病的传播风险。猫栉首蚤、犬栉首蚤等为海南地区犬类常见体表寄生蚤,它们的叮咬可引发跳蚤过敏性皮炎(FAD)、宿主贫血等直接危害,严重威胁宿主健康。目前关于海南地区犬源寄生蚤的种类组成和其微生物群落的完整多样性及其携带的潜在病原体,仍缺乏系统深入的研究[12]。由于海南本土基础数据较为匮乏,因此,本研究拟结合形态学与分子生物学方法精准鉴定海南地区犬源寄生蚤种类,采用宏基因组技术全面解析其微生物群落的结构与功能,筛选鉴定潜在致病性微生物,为针对海南地区的蚤传疾病进行精准风险评估和科学防控的研究提供关键的本土数据与理论支撑。
-
高通量组织研磨机(上海析牛莱伯仪器有限公司);金属浴SDBI10(上海小聪科技有限公司);高速台式冷冻离心机(湖南湘仪实验室仪器开发有限公司);体视显微镜(深圳市奥斯微光学仪器有限公司);琼脂糖水平电泳仪(武汉赛维尔生物科技有限公司);PCR仪(上海伯乐生命医学产品有限公司);漩涡混合仪(河北越凯科技有限公司);Agilen t Bioanalyzer 2100生物分析仪(上海欧易生物医学科技有限公司);Qubit 荧光计精准核酸定量仪(赛默飞世尔科技(中国)有限公司);NanoDrop 2000分光光度计(赛默飞世尔科技(中国)有限公司);S220聚焦超声仪(柏莱源(天津)生物科技有限公司);Agencourt AMPure XP磁珠(赛默飞世尔科技(中国)有限公司);TruSeq Nano DNA LT Sample Preparation Kit(Illumina, San Diego, CA, USA)
-
Ezup柱式动物基因组DNA抽提试剂盒(上海生工生物工程技术服务有限公司)、SanPrep柱式DNA胶回收试剂盒(上海生工生物工程技术服务有限公司)、DL 2000 DNA Marker(上海生工生物工程技术服务有限公司)、Goldview染料(北京赛百盛基因技术有限公司)、2X Taq Plus Master Mix II(Dye Plus)(南京诺维赞生物科技股份有限公司)、二甲苯(西陇科学股份有限公司)、乙醇(西陇科学股份有限公司)、氢氧化钾(西陇科学股份有限公司)、MagPure Soil DNA LQ Kit(广州美基生物科技有限公司)、VAHTS Universal PLUS DNA Library Prep Kit for Illumina®(南京诺维赞生物科技股份有限公司)、Qubit dsDNA Assay Kit(赛默飞世尔科技(中国)有限公司)、Bioanalyzer
2100 DNA-1000 Kit(安捷伦科技有限公司)。 -
2025年5—7月,在海南省三个地区采集跳蚤样本。采集地分别位于北部海口市(HK;北纬20°02′,东经110°19′)、东部文昌市(WC;北纬19°37′,东经110°45′)和中部定安县(DA;北纬19°42′,东经110°21′)。采用逆毛梳刷法配合精细镊子采集蚤样本,干冰保存运输至实验室,并置于−20°C冰箱保存。之后对样本进行分组,将每个地区的跳蚤标本进一步平均分配并构建为4个独立的生物学重复组(每个地区4个生物学重复)。最终,共获得12个样本组,用于后续的宏基因组测序与比较分析。参照《中国重要医学昆虫分类鉴定》[13]中描述的鉴定标准,通过体视显微镜对蚤标本进行形态学鉴定。从每个地区选取3只蚤类进行物种鉴定。使用Ezup柱式动物基因组DNA纯化试剂盒提取DNA,对提取的DNA的COI基因进行PCR扩增,使用1%琼脂糖凝胶电泳检测,将胶回收产物送至生工生物公司测序,测序结果与NCBI数据库比对,最终确定跳蚤的种类。
-
基于线粒体细胞色素C氧化酶亚基I(COI)基因序列构建系统发育树。从GenBank中检索并纳入了代表9个不同属(角叶蚤属、鬃蚤属、病蚤属、细蚤属、副角蚤属、栉首蚤属、蚤属、冠蚤属、客蚤属)的11条蚤COI序列。家蚕(GenBank登录号:HQ923134.1)的COI序列被用作外群以确定树的根。使用MAFFT算法对COI基因进行多序列比对。系统发育树采用邻接法(Neighbor-Joining, NJ)和p-distance模型,在MEGA11软件中重建。通过1 000次自举重复评估树拓扑结构的稳健性。使用iTOL(Interactive Tree of Life iTOL)v6在线工具对生成的系统发育树进行可视化和注释[14]。
-
采用宏基因组测序方法,对猫栉首蚤样本中的微生物的基因组DNA进行测序。使用QIAamp® Fast DNA Stool Mini Kit(Qiagen, Hilden, Germany)提取蚤DNA。随后使用NanoDrop 2000分光光度计和琼脂糖凝胶电泳评估提取DNA的浓度和完整性。通过质量控制的DNA样本使用S220聚焦超声仪随机打断,并使用Agencourt AMPure XP磁珠进行纯化。使用TruSeq Nano DNA LT Sample Preparation Kit制备测序文库,并在Illumina NovaSeq 6000平台上进行测序,生成150 bp的双端读长。本部分的文库制备、测序及后续生物信息学分析由上海欧易生物科技有限公司完成。
-
使用fastp(v0.20.1)对原始FastQ文件进行质控,包括去除接头、过滤低质量碱基及丢弃含有模糊核苷酸(N)的读长。然后,使用bbmap(v38.93-0)将质量过滤后的读长与宿主基因组进行比对,以去除宿主来源的序列,得到高质量、无宿主污染的读长,然后使用MEGAHIT(v1.2.9)进行从头组装成支架。使用Prodigal(v2.6.3)预测组装支架中的开放阅读框(ORFs),并翻译为氨基酸序列。使用MMseqs2(v13.45111),设定参数为95%序列一致性和90%覆盖度,将所有样本预测的基因进行聚类,构建非冗余基因目录。每个聚类中最长的基因被选为代表序列。使用Salmon(v1.8.0)将每个样本的洁净读长回帖到此非冗余基因集(一致性阈值设为95%)以量化基因丰度。对于分类学注释,基于NR数据库的分类信息对基因进行注释,并通过汇总归属于同一物种的所有基因的丰度来计算物种丰度。使用DIAMOND(v0.9.10.111)将代表性氨基酸序列与KEGG(Kyoto Encyclopedia of Genes and Genomes KEGG)数据库进行比对以获得KEGG通路信息,从而实现功能注释。
-
本实验所有统计分析和可视化都在R环境(v4.1.2)中进行,包括基于基因、物种和功能丰度数据的主坐标分析PCoA(Principal Coordinates Analysis PCoA)、物种和功能丰度的描述性与比较分析。采用单因素方差分析(one-way ANOVA)进行组间比较,随后进行Tukey事后检验。以P<0.05为差异具有统计学意义。
-
本研究共从120只犬体表采集到的319只蚤类(209只雌性,110只雄性)。各地区样本数量及占比分别为海口115只(36%)、定安99只(31%)和文昌105只(33%)。经鉴定蚤类样本的形态结构与猫栉首蚤(Ctenocephalides felis)形态特征基本一致,初步判断为猫栉首蚤。猫栉首蚤外观呈棕黄色,具有刺吸式口器,虫体细小,且左右扁平、无翅、有3对发达的足;猫栉首蚤具有坚硬的外骨骼,体壁硬而光滑(图1)。分子生物学鉴定显示,在凝胶图上约610 bp处可见明显条带(图S1)。经测序结果显示与猫栉首蚤凭证标本IZ08_1单倍型Cf_h3(GenBank 登录号: MW136216.1)的CO1基因一致性达99.7%,从而确认蚤样本为猫栉首蚤。
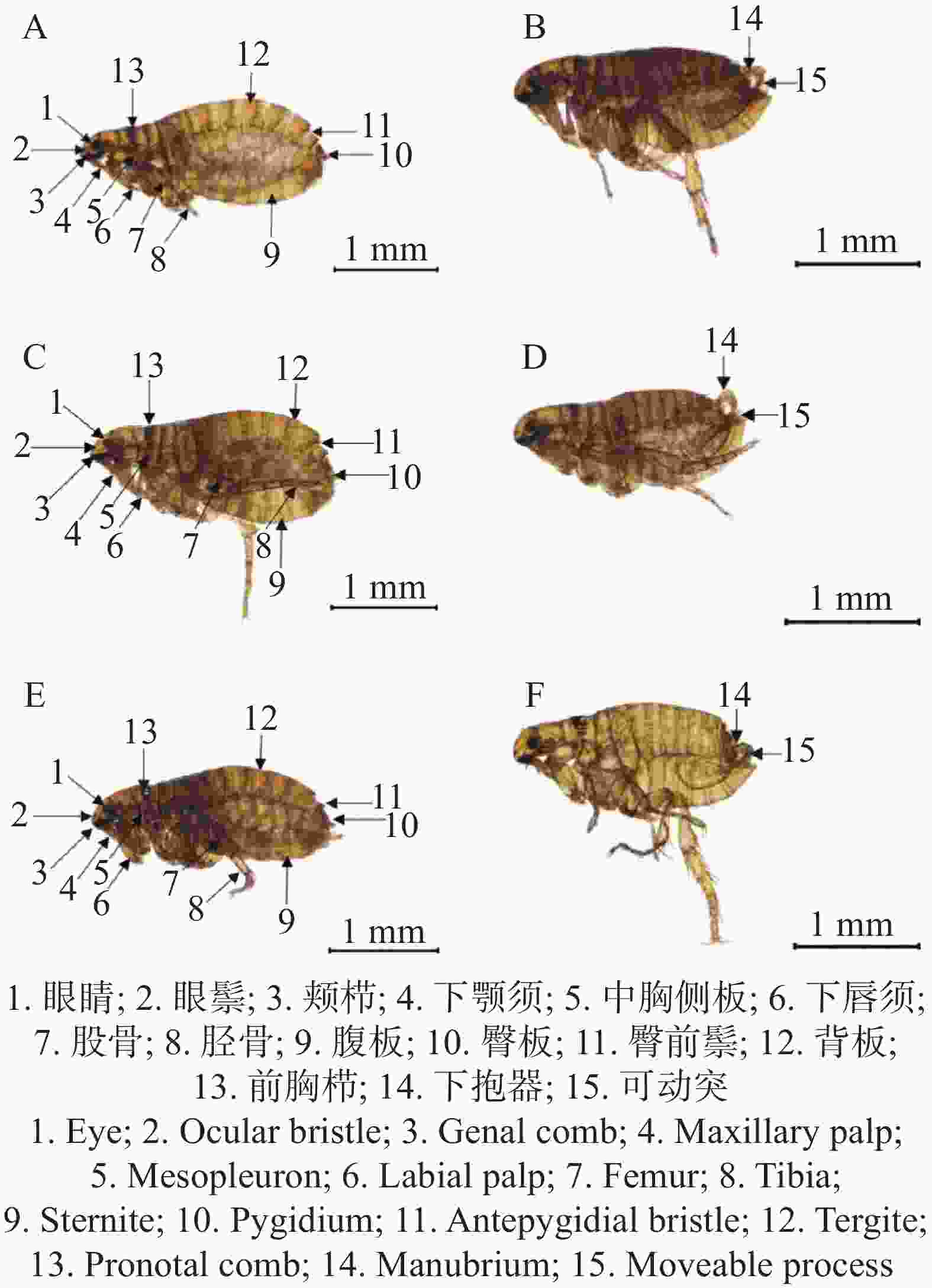
Figure 1. Morphological identification of C. felis in Hainan region
-
在蚤类进化支内,角叶蚤属和鬃蚤属形成姐妹群。来自海南的猫栉首蚤(MW136216.1)与其同属物种犬栉首蚤以高自举支持率(100%)紧密聚在一起,形成一个独立的分支(图2)。
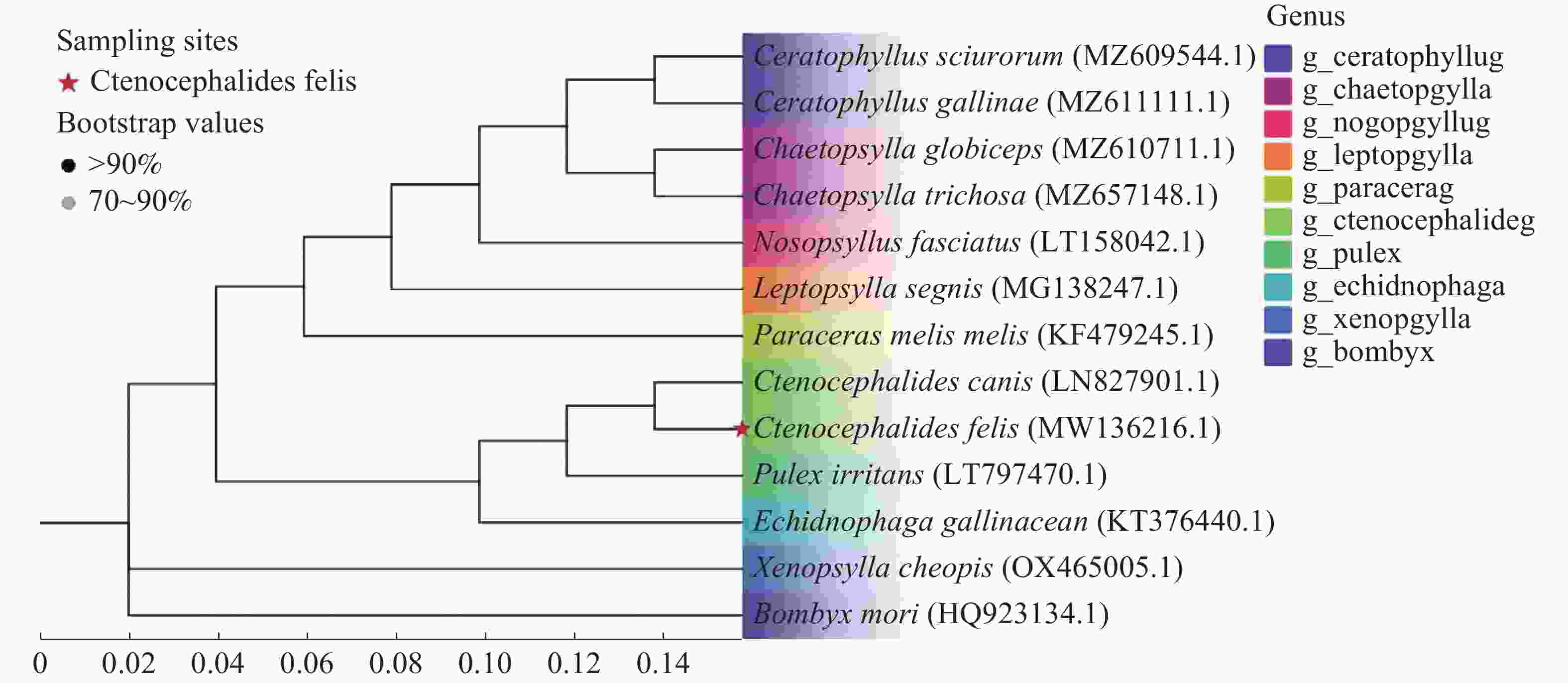
Figure 2. Neighbor-Joining phylogenetic tree of fleas based on mitochondrial COI gene sequences
-
宏基因测序共得到原始数据34.07 G,有效数据33.33 G,数据有效率为97.84%,GC占比30.20%,Q30占比97.23%,Q20占比99.48%(表1)。
样本编号 原始数据/G 有效数据/G 有效数据率/% GC/% Q30/% Q20/% HK 11.27 11.02 97.81% 30.32% 97.22% 99.47% DA 11.43 11.17 97.71% 30.15% 97.23% 99.49% WC 11.37 11.14 97.99% 30.12% 97.24% 99.47% 注:HK、DA和WC分别表示海口、定安和文昌的猫栉首蚤样本。有效数据比率表示有效测序序列数占原始测序序列数的比例;GC表示碱基G和C数量之和占总碱基数量的百分比;Q30表示质量>30的碱基占总数据量的比例;Q20表示质量>20的碱基占总数据量的比例。 Note: HK, DA and WC represent the C. felis samples collected from Haikou, Ding'an and Wenchang, respectively. The valid data ratio refers to the proportion of valid sequencing reads to the total raw sequencing reads; GC content denotes the percentage of the sum of guanine(G)and cytosine(C)bases relative to the total number of bases; Q30 indicates the proportion of bases with a quality score >30 relative to the total data volume; Q20 represents the proportion of bases with a quality score >20 relative to the total data volume. Table 1. Metagenomic sequencing data of C. felis in Hainan region
-
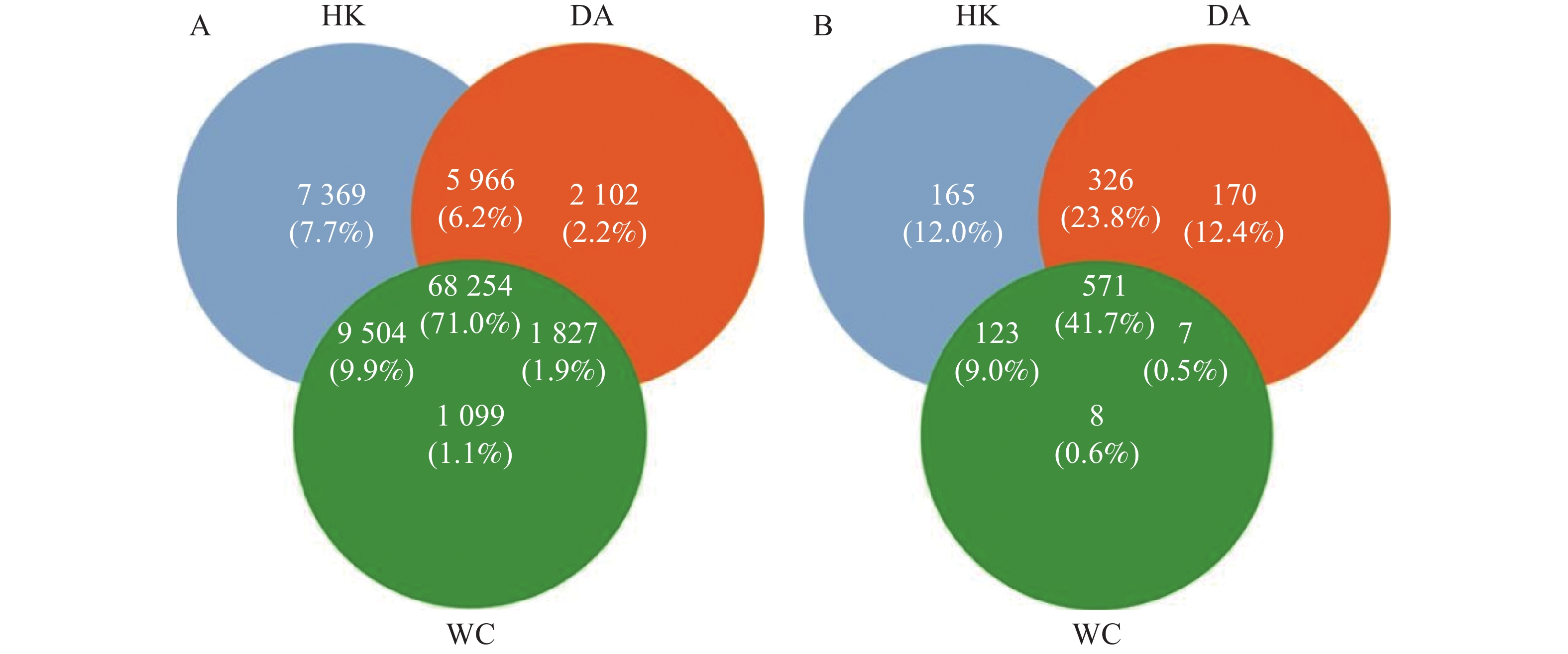
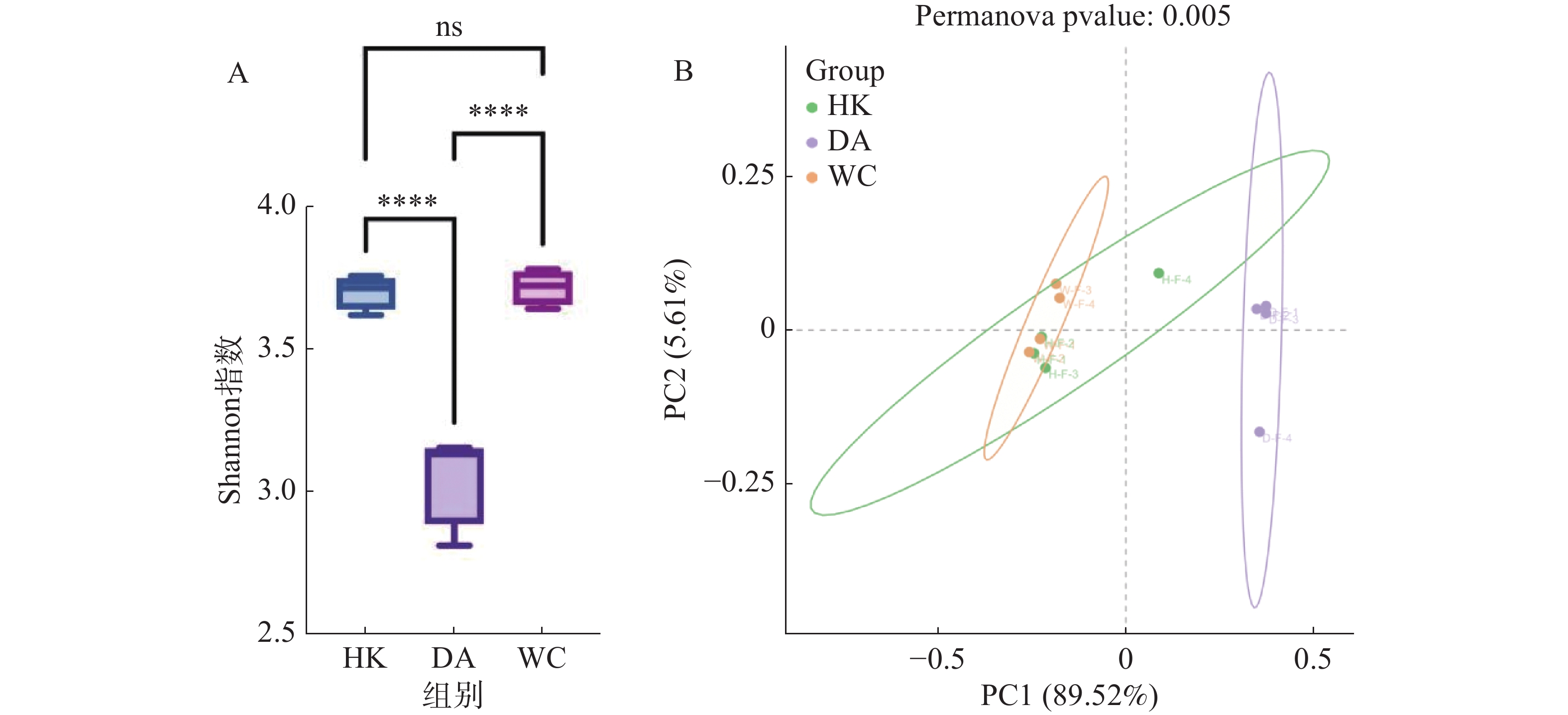
从基因水平(图3)可以看出,3个样本共含有68 254个基因。依数量大小排名为HK>DA>WC,HK、DA、WC独有的基因数量分别为7 369、2 102和1 099。从种水平(图3-B)可以看出,三个样本含有共同物种为571个,依丰富度大小排名为DA>HK>WC,三个样本独有的种数量分别为165、170和8。多样性分析显示(图4-A),三组样本整体存在显著差异(F=49.43,R2=0.916,P<0.001)。WC和HK样本微生物群落丰度显著高于DA样本,其中WC样本的微生物群落多样性最高,HK与WC样本无显著差异(P>0.05)。基于Bray-Curtis距离的主坐标分析图显示(图4-B),PC1和PC2对样本间微生物物种变异的贡献率分别为89.52%和5.61%。HK与DA、WC样本有交叉重叠区域,这表明它们在微生物群落结构上具有较高的相似性。相比之下DA样本和WC样本距离较远,菌群差异明显,能够单独聚类,表明DA样本微生物群落结构与HK、WC样本存在显著差异(P<0.05)。
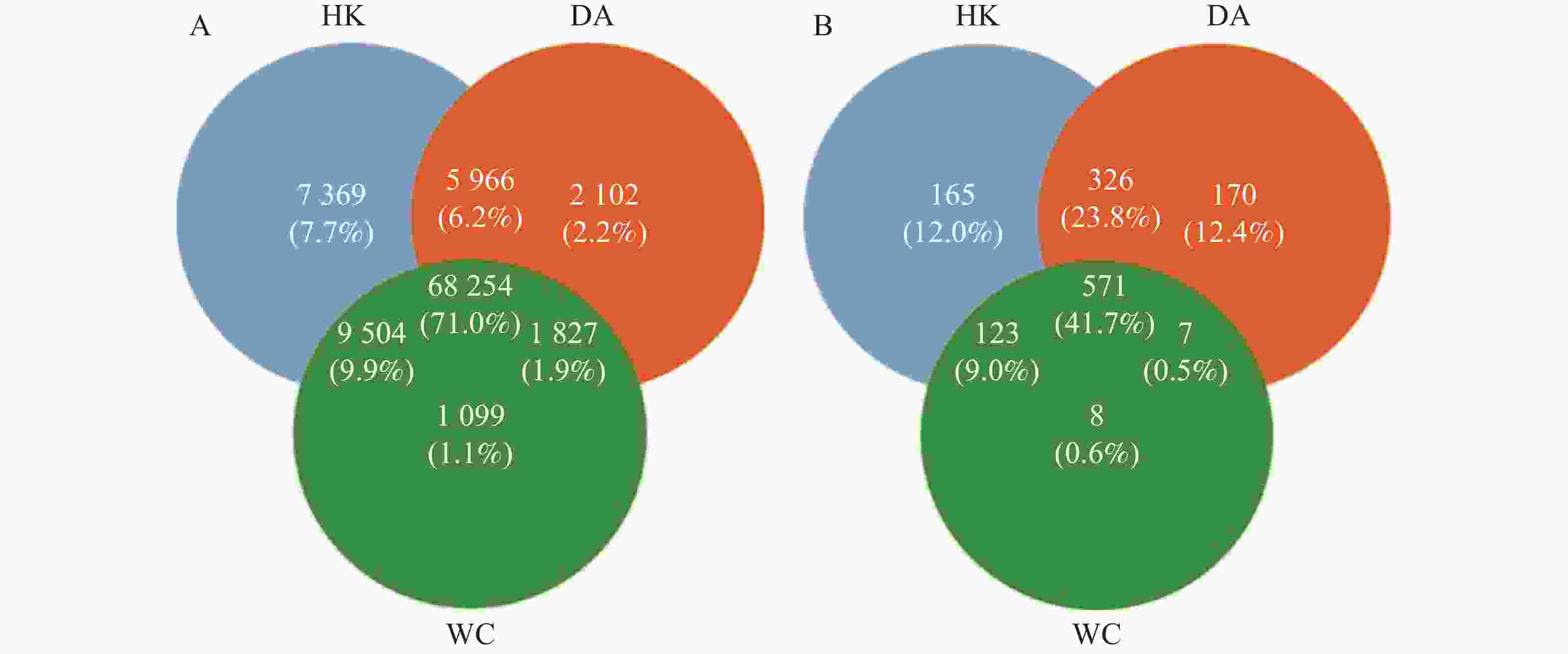
Figure 3. Venn diagram of bacteria carried by C. felis in Hainan region
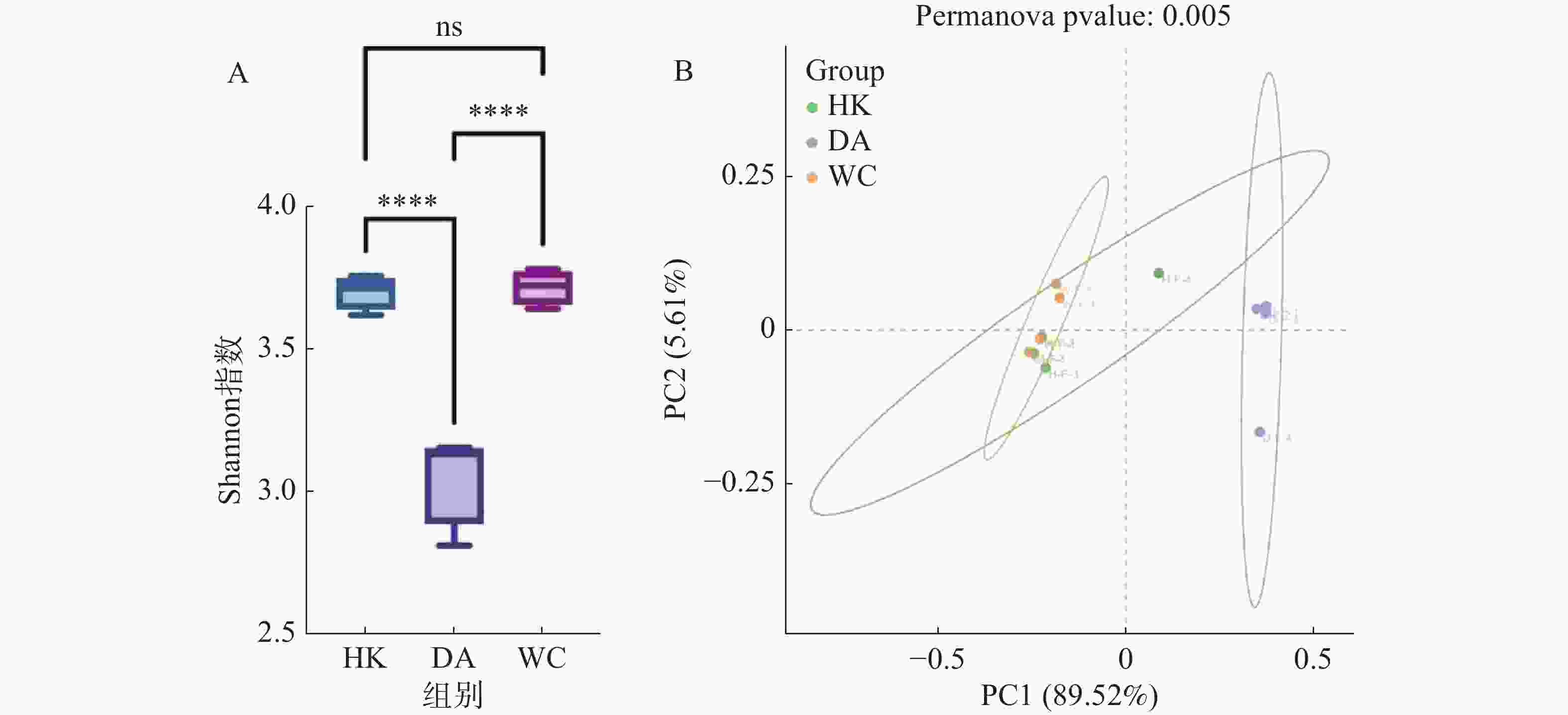
Figure 4. Differential analysis of microorganisms carried by C. felis in Hainan region
-
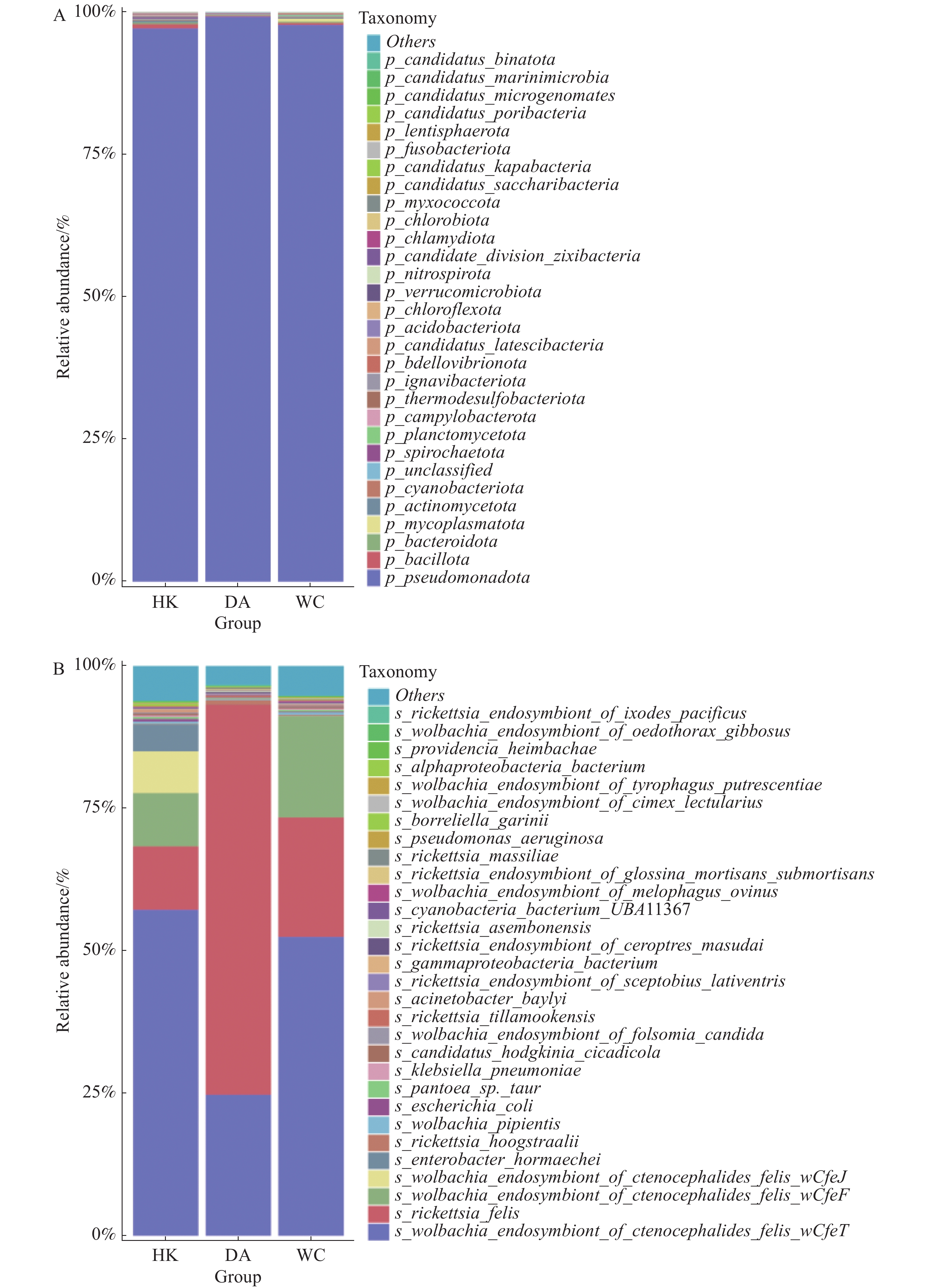
在门水平上(图5-A),假单胞菌门(Pseudomonadota)为三个样本的优势菌门。厚壁菌门(Bacillota)为HK和DA第二大优势门,而支原体门(Mycoplasmatota)为WC的第二优势门。在种水平上共注释到788个细菌。相对丰度排名前30的物种箱线图见图5-B。在三个地区中,相对丰度排名前五的物种一致,依次为猫栉首蚤沃尔巴克氏体内共生体wOfeT株(Wolbachia endosymbiont of Ctenocephalides felis wOfeT)、猫立克次体(Rickettsia felis)、猫栉首蚤沃尔巴克氏体内共生体wCfeF株(Wolbachia endosymbiont of Ctenocephalides felis wCfeF)、猫栉首蚤沃尔巴克氏体内共生体wOfeJ株(Wolbachia endosymbiont of Ctenocephalides felis wOfeJ)及霍氏肠杆菌(Enterobacter hormaechei)。其中,能导致人兽共患细菌有猫立克次体(Rickettsia. felis)、肺炎克雷伯菌(Klebsiella. pneumoniae)、蒂拉穆克立克次体(Rickettsia. tillamookensiss)、马赛立克次体(Rickettsia. massiliae)、铜绿假单胞菌(Pseudomonas. aeruginosa)和伽氏疏螺旋体(Borreliella garinii)。其中,猫立克次体丰度最高,占比也最高,占总检测物种的36.6%,DA、WC和HK猫立克次体丰度占比分别为68.5%、20.9%和11.2%。此外猫立克次体可以引发猫抓热、鼠伤寒、蚤传斑疹热和鼠疫等疾病[15−16],给动物和人类健康造成极大的危害。其余物种,如霍氏肠杆菌(Enterobacter hormaechei)、海姆巴赫普罗维登斯菌(Providencia heimbachae)、贝氏不动杆菌(Acinetobacter baylyi)等为条件致病菌,主要与动物或人类的肠道疾病有关。
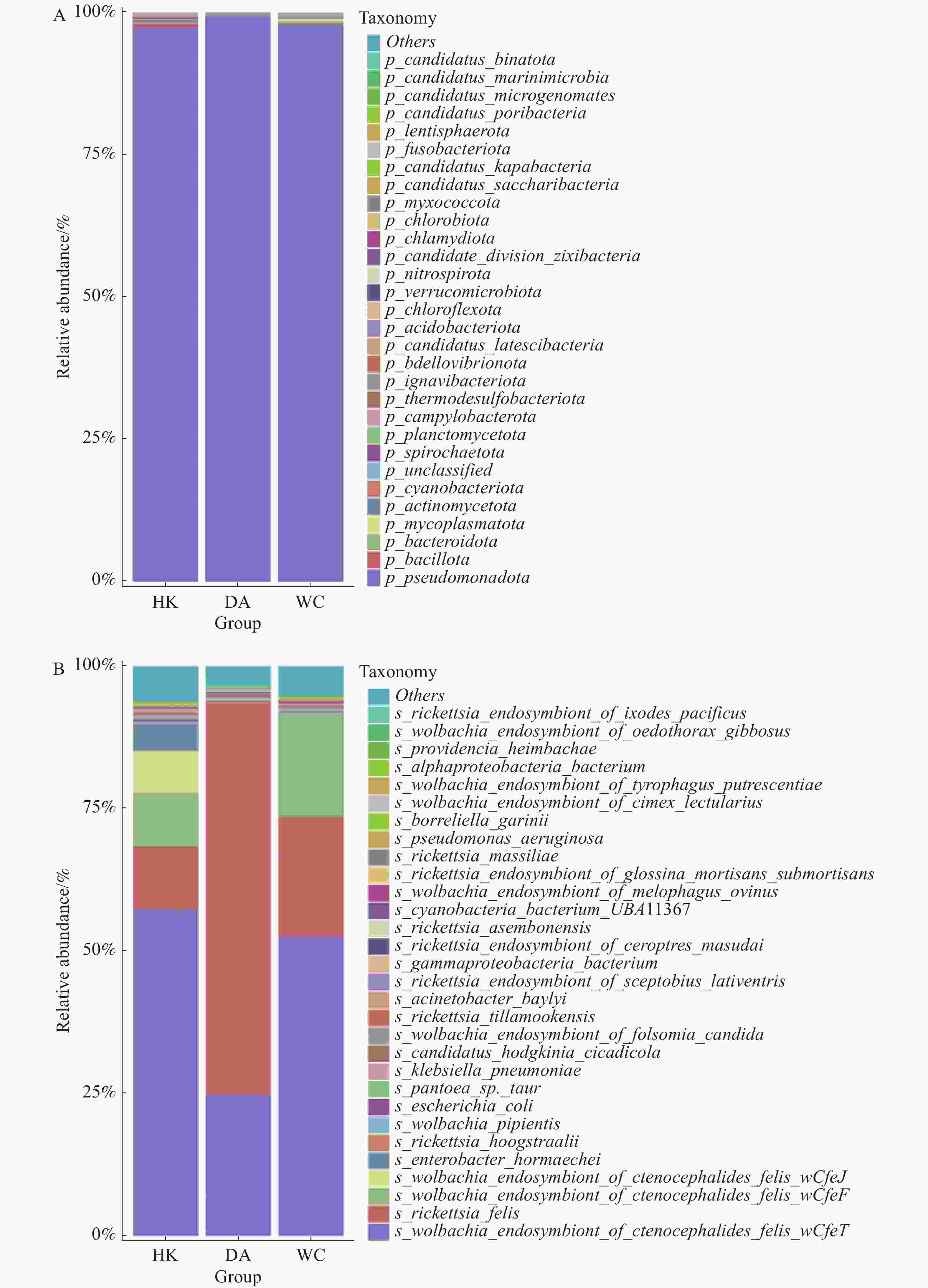
Figure 5. Relative abundance of bacteria at the phylum and species levels carried by C. felis in Hainan region
-
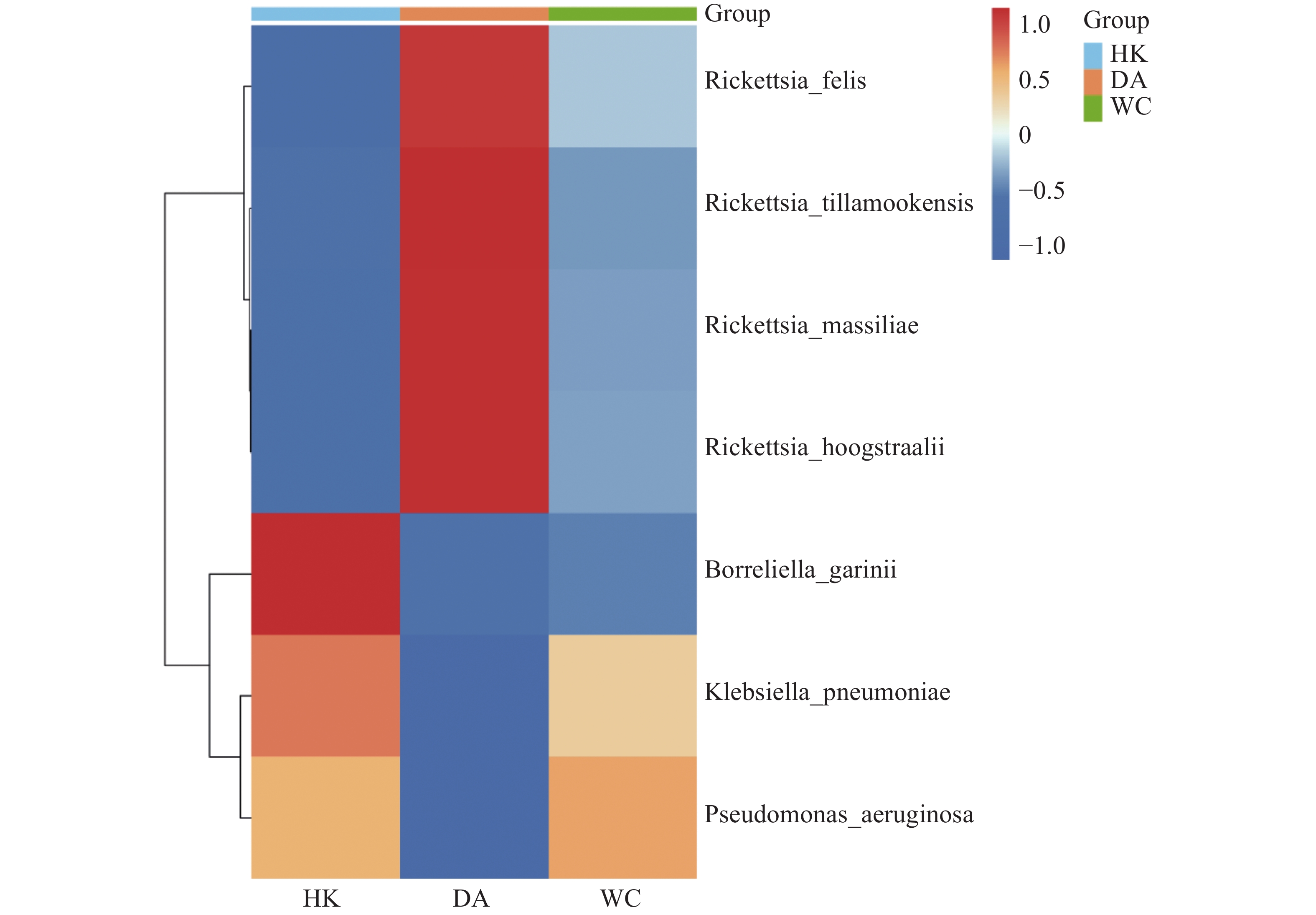
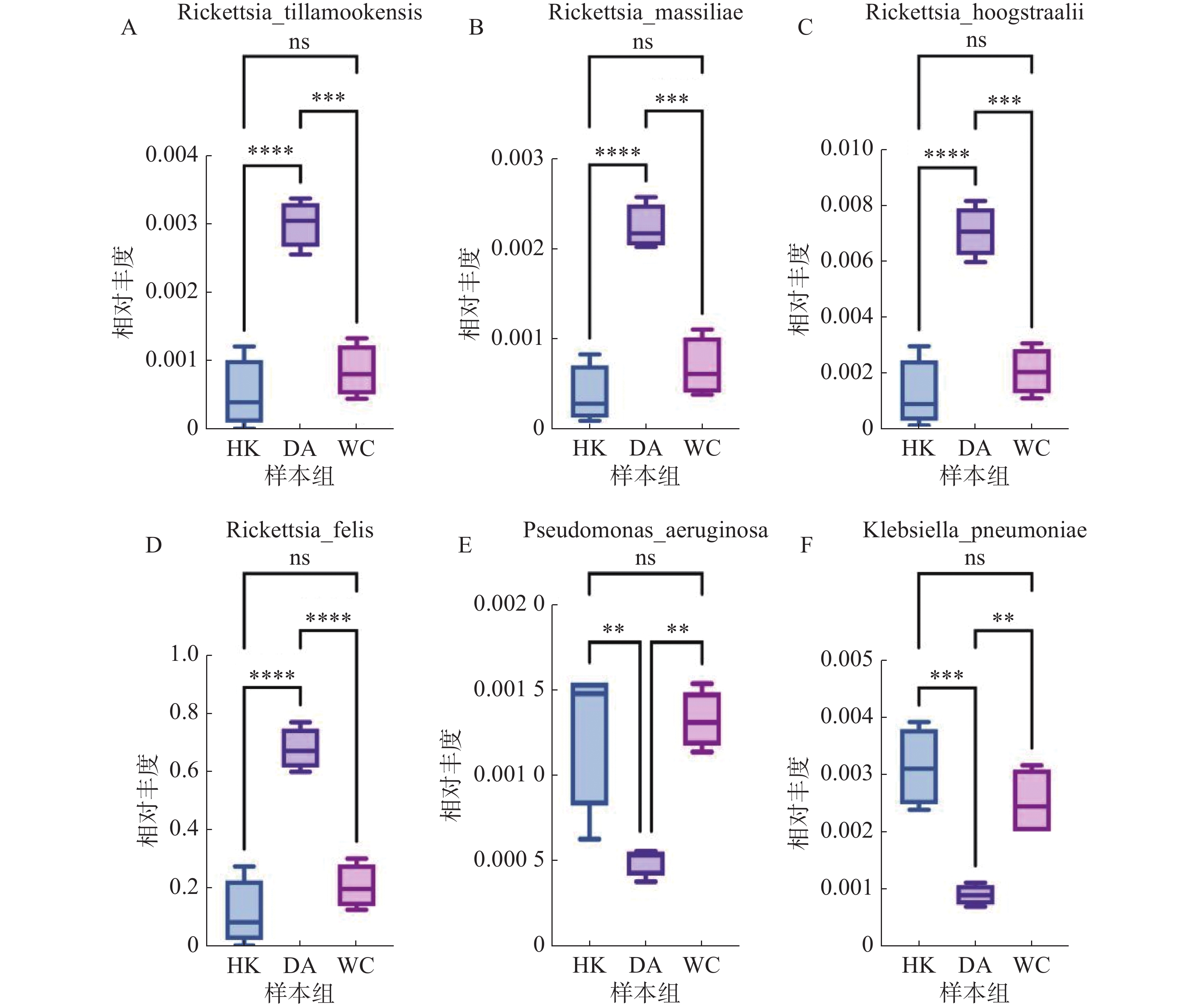
根据致病菌热图分析结果(图6)显示,立克次体属主要富集在DA样本;伽氏疏螺旋体(Borreliella garinii)主要富集在HK样本中;肺炎克雷伯菌(Klebsiella. pneumoniae)和霍氏立克次体(Rickettsia. hoogstraalii)主要富集在HK和WC样本。分析结果显示,立克次体属(R2=0.910)(图7 A-D)HK样本丰度显著低于DA和WC样本;蒂拉穆克立克次体(Rickettsia. tillamookensiss)(R2=0.904)、马赛立克次体(Rickettsia. massiliae)(R2=
0.9083 )、霍氏立克次体(Rickettsia. hoogstraalii)(R2=0.900)和猫立克次体(Rickettsia. felis)(R2=0.910)在DA与HK、DA与WC存在显著差异(P<0.001)。分析结果(图7-E—F)显示,HK样本丰度显著高于DA和WC样本,其中铜绿假单胞菌(Pseudomonas. aeruginosa)(R2=0.714)和肺炎克雷伯菌(Klebsiella. pneumoniae)(R2=0.808)在DA与HK、DA与WC存在显著差异(P<0.05)。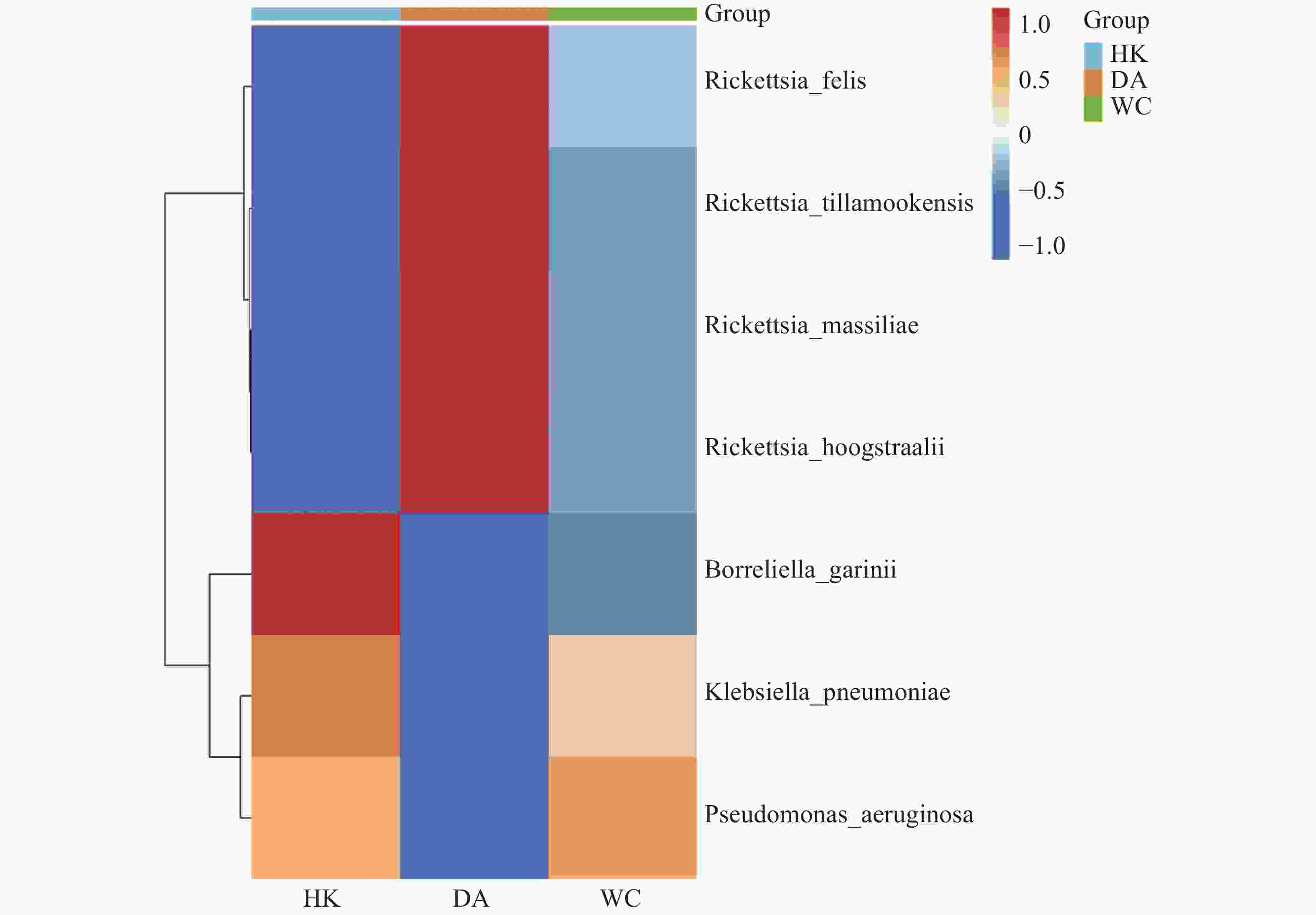
Figure 6. Heatmap of pathogens carried by C. felis in Hainan
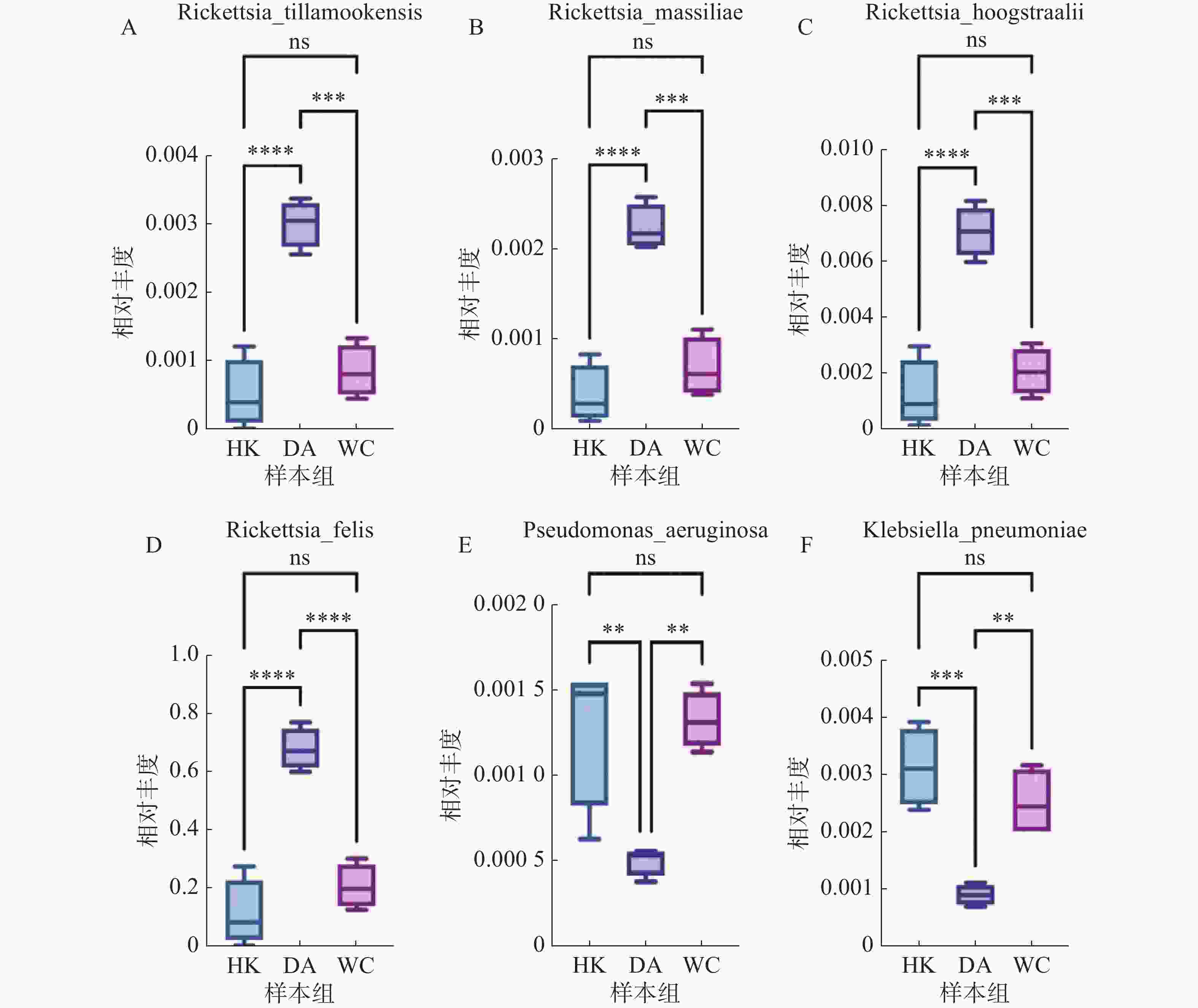
Figure 7. Diversity analysis of pathogenic bacteria carried by C. felis in Hainan region
-
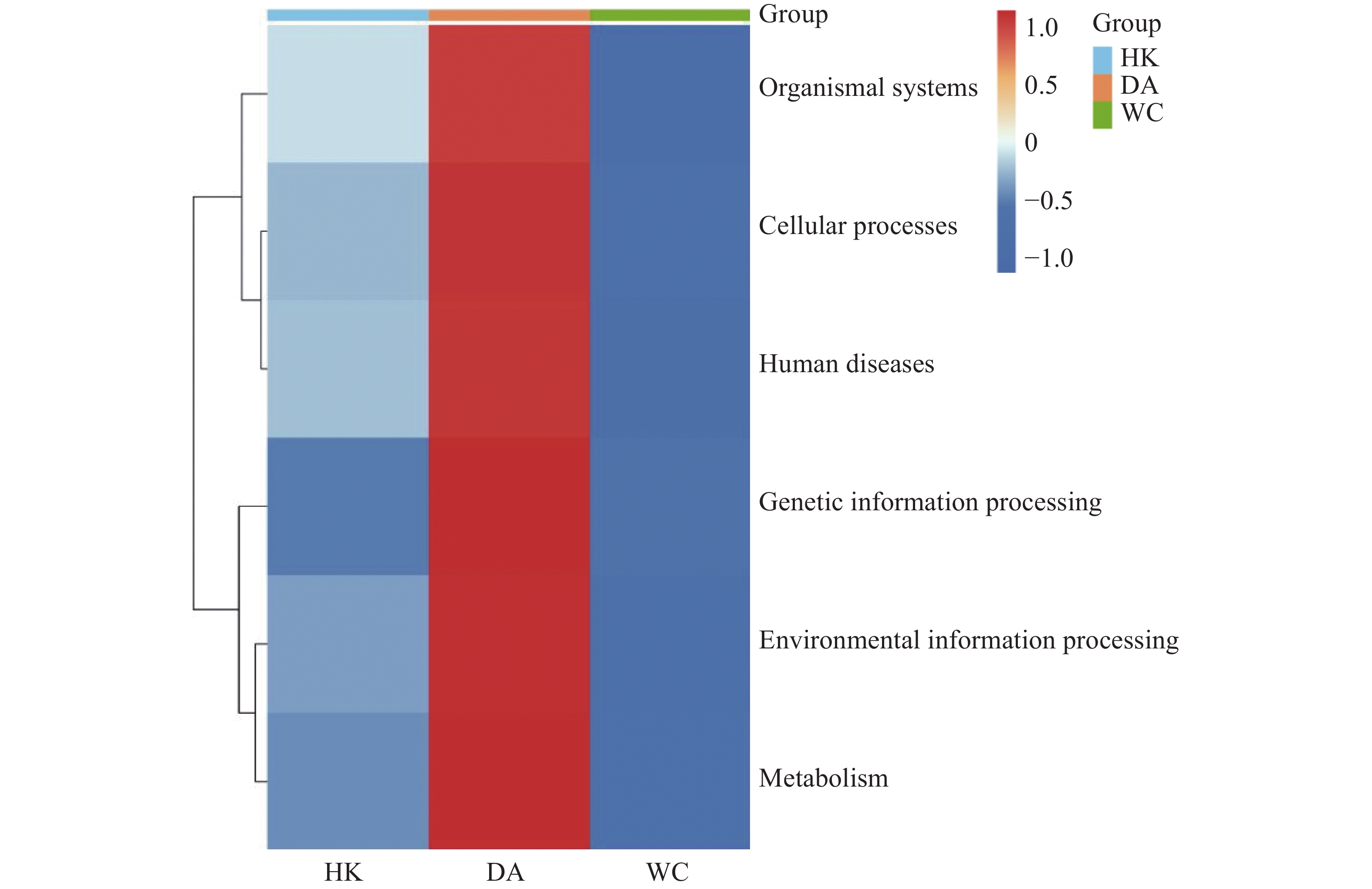
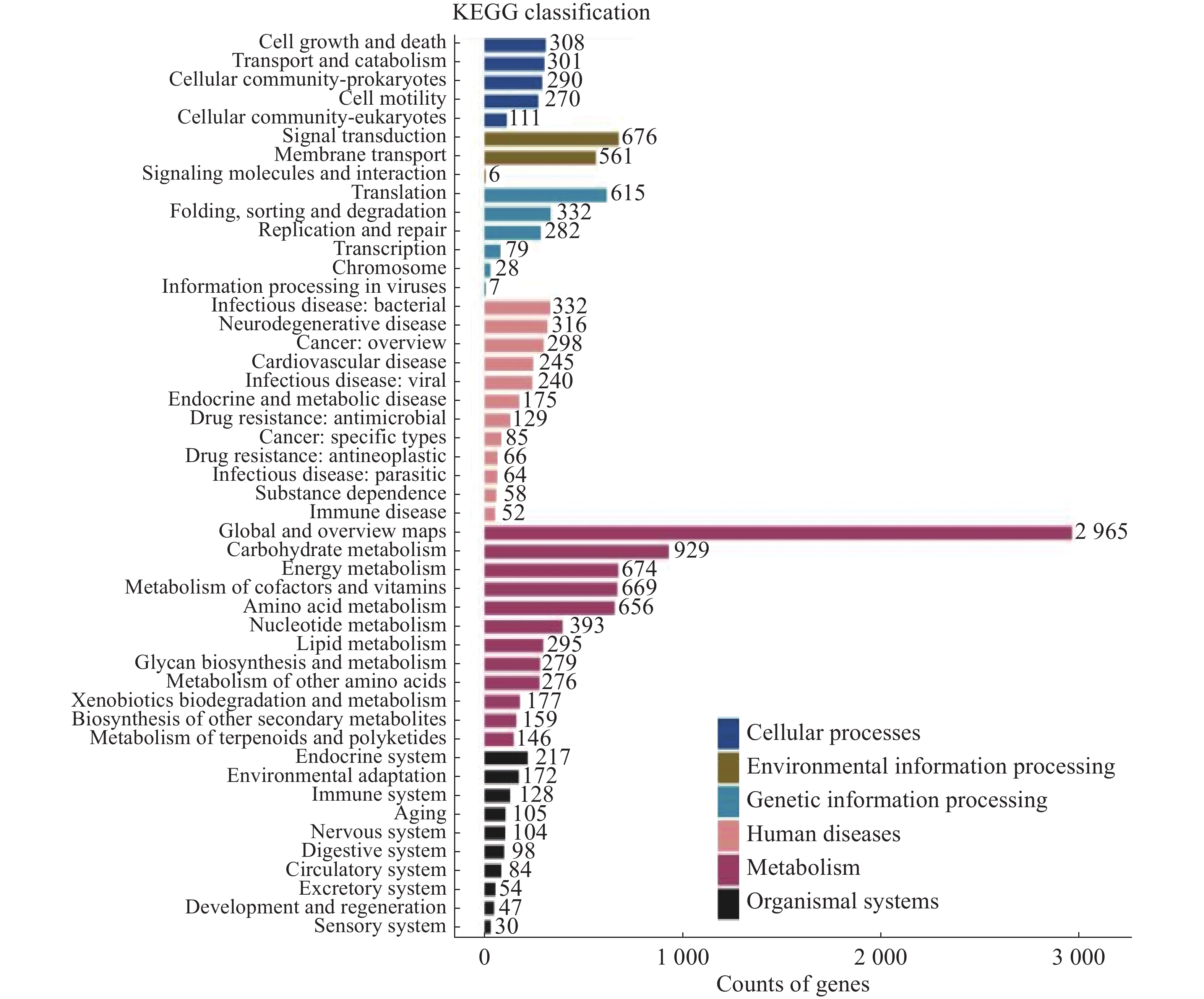
将14 104个非冗余基因与KEGG数据库进行比对。KEGG数据库注释到6个一级代谢通路,分别为有机系统(Organismal Systems)、细胞过程(Cellular Processes)、人类疾病(Human Diseases)、遗传信息处理(Genetic Information Processing)、环境信息处理(Environmental Information Processing)和代谢(Metabolism)通路。基因功能注释热图(图8)显示DA样本的6个一级通路相较于其他样本相对丰度最高,其中细胞过程、遗传信息处理和代谢通路最高;HK和WC各通路的相对丰度水平类似。KEGG数据库注释的二级通路相对应的基因数量图(图9)显示,一共可以注释到48个二级通路,在一级通路中,代谢和人类疾病通路含有的二级通路最多,为12个;代谢通路含有的基因数量最多,有7 618个基因;环境信息处理通路含有的二级通路最少,相应的基因数量也最少,分别为3个二级通路和1 243个基因。
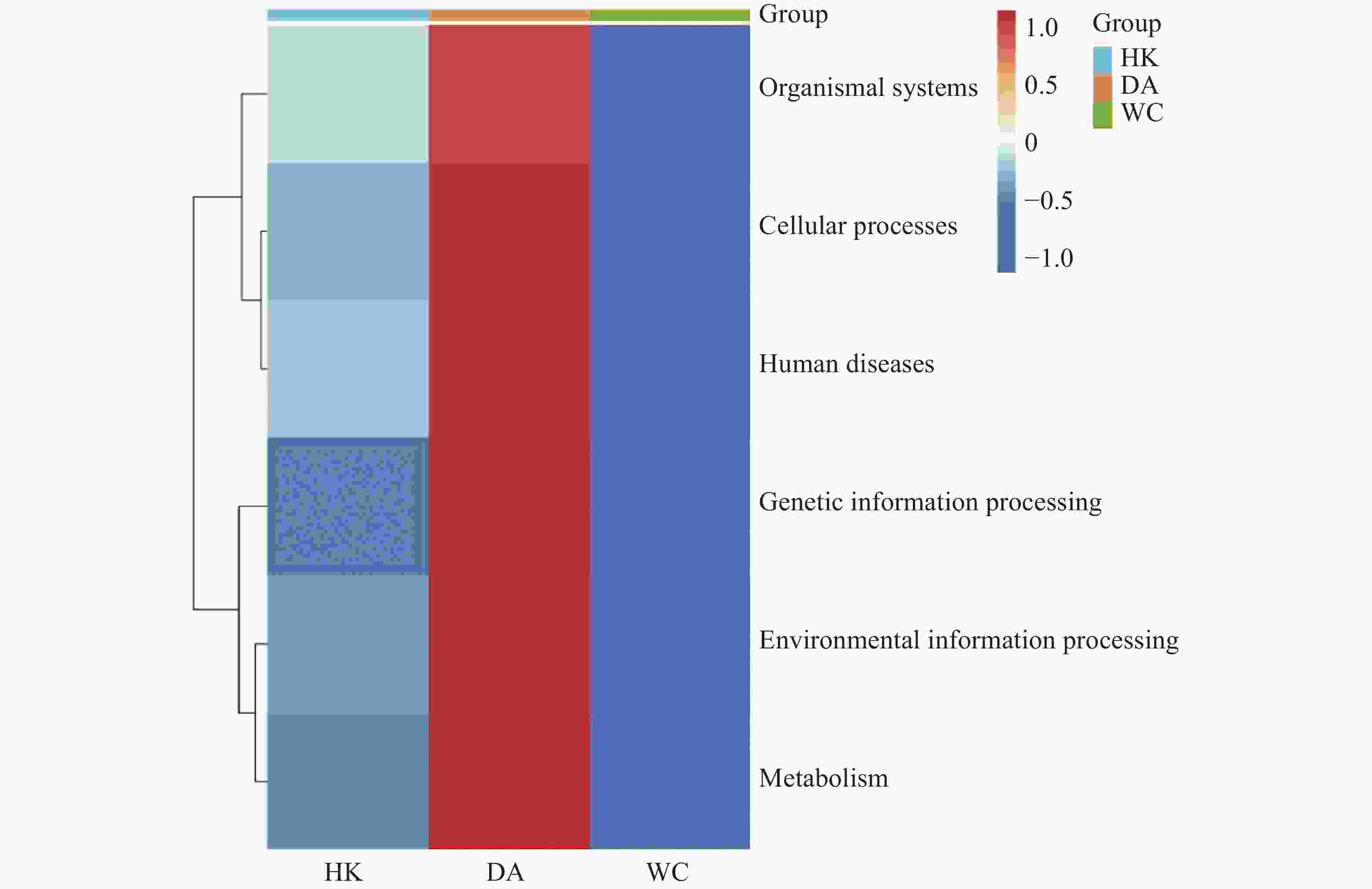
Figure 8. Heatmap of functional annotation of bacterial genes from C. felis in Hainan region
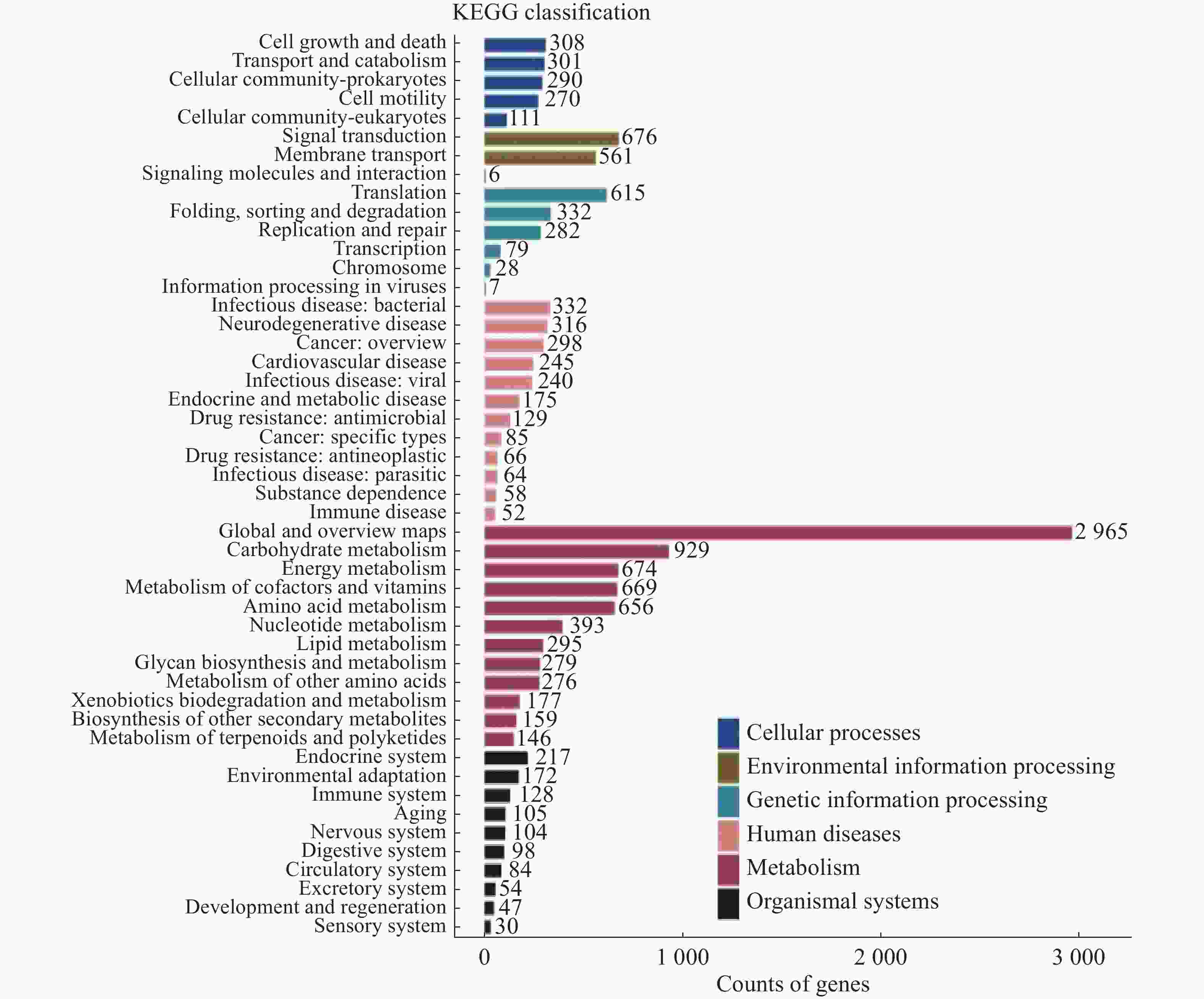
Figure 9. Statistics of annotated gene numbers in metabolic pathways of bacteria from C. felis in Hainan region
-
本研究揭示假单胞菌门为猫栉首蚤的最大细菌门,包含众多病原体,也是本研究中测序鉴定出的最丰富的细菌门。这一发现与其他媒介物种中的研究结果一致,例如鸡皮刺螨[17]、翅螨科和巨刺螨科的成员[18]、鼻刺螨科,这表明大多数假单胞菌门细菌在节肢动物中广泛分布。假单胞菌门细菌可能参与调节宿主昆虫肠道的多种生理功能,包括代谢途径和消化过程[19−20],这提示其在猫栉首蚤代谢中可能发挥关键作用。在昆虫的肠道微生物群落中,厚壁菌门是主要的组成部分之一,可以协助宿主昆虫进行营养代谢,提供食物中缺乏的养分,促进昆虫的生长和繁殖[21],同时厚壁菌门的细菌能通过分泌抗菌肽[22]、毒素等增强昆虫对寄生物的防御能力和抗病性。
本研究在猫栉首蚤中检测到猫立克次体、蒂拉穆克立克次体等多种致病菌,进一步佐证了立克次体在节肢动物中的广泛分布。已有多项研究为此提供了支持:1)SALCEDA‐SáNCHEZ 等[23]通过常规PCR扩增与测序技术,在蚤类中成功鉴定出猫立克次体;2)HORTA M C等人[24]借助PCR检测,在巴西的猫栉首蚤中发现了立克次体感染;3)RADZIJEVSKAJA J等人[25]对立陶宛啮齿类动物体外寄生虫的立克次体感染情况展开调查,结果显示蚤类的感染率最高,且涉及四种立克次体物种。研究结果表明地理位置可能是显著影响猫栉首蚤的微生物群及其基因表达的关键因素。核心因素可能是由各区域的自然地理气候条件、犬只的种群特征及饲养管理模式、城市化程度及人类生产生活活动的差异导致的,同时也受微生物与猫栉首蚤共生传播特性的影响;探究地域差异,不仅能丰富热带地区媒介生物—宿主—微生物共生的生态学研究数据,填补海南岛相关研究的不足,还能精准识别海南蚤传人兽共患病的风险分布特征,为当地管理部门制定差异化的蚤类及蚤传疾病防控策略提供科学依据,也为中国华南热带地区及其他热带岛屿的同类媒介生物防控与公共卫生实践提供参考。不同地区猫栉首蚤的微生物群落存在显著差异,本研究中WC与HK样本的微生物群落丰度显著高于DA样本,但DA样本含有的致病菌丰富最高,这一差异可能与地理位置的影响密切相关。对KEGG数据的统计分析结果表明,主要的细菌基因与代谢途径相关,通过协助昆虫分解和吸收食物中的营养物质来促进生长和繁殖[26]。代谢通路包括的二级通路有碳水化合物、氨基酸和脂质代谢及辅因子和维生素的代谢,这些与猫栉首蚤的营养获取、能量产生、代谢及自噬—溶酶体信号通路密切相关。这些代谢途径通过影响生长、繁殖和抵御病原体,在生理过程中发挥着关键作用,这与RIVERA-PéREZ C等[27]的研究发现一致,他们证实代谢途径可通过调控营养获取与能量产生,对蚊子的生理功能产生决定性影响。作为多种疾病的传播媒介,猫栉首蚤的肠道微生物群可能通过调控病原体增殖及传播相关基因表达,直接参与疾病传播过程。这一推测得到了同类研究的支持:GAO H等[28]的研究证实,蚊子肠道微生物群可显著影响疾病传播效率,其核心机制与本研究揭示的节肢动物肠道微生物群介导病原体传播的结论高度一致,进一步验证了该调控模式在吸血节肢动物中的普遍性。猫栉首蚤通过吸食受感染宿主的血液获得病原体,其肠道微生物群的组成和稳定性直接决定了其对病原体的易感性和传播能力。DENNISON N J等[29]在蚊子中证明了相同的机制,即肠道微生物群的组成直接影响对病原体的易感性和传播效率。这些发现共同表明,猫栉首蚤的肠道微生物群不仅参与代谢过程,还影响人类疾病的传播,从而为控制蚤传疾病确定了新的干预靶点。
媒介生物所携带的微生物群落可能受到多种因素的影响,如蚤种、地理位置和气候等因素影响细菌群落的组成[30]。这些发现表明,媒介生物携带的微生物群是由复杂的相互作用因素影响的,并且对不同媒介物种的影响各不相同。栖息地越相似,共生微生物群落的相似性就越大。
本研究揭示了猫栉首蚤的微生物群落结构及其功能特征。研究结果表明,地理因素是影响其微生物组成的重要因素。值得注意的是,本研究以猫栉首蚤为研究对象,鉴定出多种对哺乳动物健康构成潜在风险的致病菌,提示这些病原体应在控制策略中予以优先关注。进一步的功能注释分析表明,微生物在代谢和人类疾病相关通路中富集,提示这些功能可能直接影响蚤的生理及其传播疾病的能力。本研究系统描述了海南地区猫栉首蚤的微生物群落特征,为蚤传疾病的靶向控制策略提供了重要见解。
-
本研究明确猫栉首蚤为海南地区犬体表的优势寄生蚤种,其携带高丰度致病性微生物,这提示该地区存在较高的蚤传人兽共患病潜在流行风险;猫栉首蚤微生物群落的显著地域差异,反映了海南不同区域生态因子对蚤媒微生物群落的调控作用;而微生物基因在代谢及人类疾病通路的富集,揭示了其对蚤类生理适配与病原传播的潜在功能关联。本研究为海南地区蚤传疾病的靶向监测与防控策略制定提供了科学依据。
Microbial diversity of Ctenocephalides felis from canine hosts in Hainan
DOI: 10.15886/j.cnki.rdswxb.20260007
- Received Date: 2026-01-09
- Accepted Date: 2026-02-09
- Rev Recd Date: 2026-01-29
-
Key words:
- Dog /
- Ctenocephalides felis /
- microorganism /
- metagenom
Abstract: An attempt was made to investigate the species of ectoparasitic fleas on the body surface of dogs and the pathogenic microorganisms they carry in Hainan to provide reliable data support for the scientific prevention and control of flea-borne diseases. Ectoparasitic fleas were collected from dogs in three regions of Hainan Province, namely Haikou (HK), Ding'an (DA), and Wenchang (WC), using the reverse hair brushing method combined with fine forceps. Flea species were identified by morphological observation and polymerase chain reaction (PCR). Metagenomic sequencing was used to analyze the microbial community composition and gene functions within the fleas. Results showed that the ectoparasitic flea species identified on the dogs in different regions of Hainan was Ctenocephalides felis. Alpha diversity analysis showed that there were significant overall differences among the three groups of samples (F=49.43, P<0.001). The WC and HK samples were significantly higher in microbial community richness than the DA samples, Among the samples the WC samples was the highest in microbial community diversity, and no significant difference was observed between the HK and WC samples (P>0.05). Beta diversity analysis showed that the three samples had overlapping regions, indicating a high degree of similarity in their microbial community structures. In contrast, the DA samples were clearly separated from the WC samples, showing distinct differences in their microbial communities, and the DA samples formed an independent cluster, indicating that their microbial community structure was significantly different from that of the WC and HK samples (P < 0.05). At the phylum level, Pseudomonadota was the dominant bacterial phylum in all three samples; Bacillota was the second most dominant phylum in the HK and DA samples; Mycoplasmatota was the second most dominant phylum in the WC samples. At the species level, several bacteria posing pathogenic threats to humans and animals were detected, including Rickettsia felis, Klebsiella pneumoniae, Rickettsia tillamookensis, Rickettsia massiliae, Pseudomonas aeruginosa, and Borreliella garinii. Conclusion The findings confirmed Ctenocephalides felis as the dominant ectoparasitic flea species on the dogs in Hainan. The high abundance of pathogenic microorganisms carried by this flea species indicates a relatively high potential risk of flea-borne zoonotic disease outbreaks in this region, providing a scientific basis for targeted surveillance and the formulation of prevention and control strategies for flea-borne diseases in Hainan.
| Citation: | Li Jie, Yu Hongxiao, Jitrawadee Intirach, Chen Ping, Meng Fengxia, Han Qian. Microbial diversity of Ctenocephalides felis from canine hosts in Hainan[J]. Journal of Tropical Biology. doi: 10.15886/j.cnki.rdswxb.20260007

|
 DownLoad:
DownLoad: