
-
埃里希体属(Ehrlichia)隶属于立克次体目(Rickettsiales)、埃里希体科(Ehrlichiaceae),是一类小型、革兰氏阴性和形态多样的专性胞内寄生菌,主要侵入白细胞进行复制生存[1],可导致人类和动物患病,在全球范围内广泛分布,对公共卫生与动物健康构成重要威胁。基于16S rRNA 基因序列分析,埃里希体属包含犬埃里希体(Ehrlichia canis)、查菲埃里希体(Ehrlichia chaffeensis)、伊氏埃里希体(Ehrlichia ewingii)、反刍动物埃里希体(Ehrlichia ruminantium)及鼠埃里希体(Ehrlichia muris)等菌种,其中多种成员具有明确的宿主特异性与地域性流行特点[2]。犬埃里希体(E. canis)最早于1935年于阿尔及利亚被发现[3],是犬单核细胞性埃里希体病(Canine monocytic ehrlichiosis, CME)的致病因子,也称为热带犬全血细胞减少症[4-5]。E. canis为革兰氏阴性菌,主要寄生于犬的单核细胞和巨噬细胞内,以包涵体的形式存在[6],其通过蜱虫传播,主要媒介为血红扇头蜱(Rhipicephalus sanguineus)[7-9]。E. canis感染犬通常表现发热和血小板减少,严重病例可出现慢性或致死性消耗性疾病[10-11]。E. canis在亚洲、非洲、欧洲及美洲等多个地区均有报道[12-15]。E. canis在过去的研究中主要在家犬中检出[16],日前发现E. canis除感染家犬外亦感染其他犬类动物,如土狼、红狐、灰狐等[17-19]。近年来,研究员在欧洲、亚洲、非洲等地均有发现在非犬科动物宿主中也检测到与E. canis高度同源的DNA序列,如猫、牛、鹿等多种动物,甚至在委内瑞拉国的人血样本中检出与E. canis的16S rRNA基因序列同源性高达99%的DNA序列[20-24]。目前有研究表明,中国多个省份的家养反刍动物感染了各种无形体属和埃立克体属病原体[29],并且在反刍动物身上寄生的蜱虫上也检测多种无形体属和埃立克体属病原体基因序列[30]。上述研究结果表明,E. canis可能突破传统宿主限制,具备跨宿主感染的能力。
海南黑山羊是海南省优良地方山羊品种,分布于海南各市县。在种质上兼具耐湿热、强适应、高抗病的特点,同时成熟周期较短[25],是海南省重要的畜牧资源。海南地处热带和亚热带地区,植被茂盛,气候适宜,硬蜱种类丰富[26-27]。血红扇头蜱为E. canis的主要媒介,其在海南省是常见蜱种之一[28]。黑山羊的饲养方式多为散养放养,在生态系统中常与犬及蜱虫共栖于相同的放牧环境,血红扇头蜱可能充当中间宿主。埃里希体属的感染,会导致羊群出现斑点热、厌食、嗜睡等症状,严重影响海南地区的畜牧经济效益。因此,检测黑山羊是否感染埃里希体属病原体,具有重要的流行病学意义。
16S rRNA基因作为原核生物的保守性分子标记,因其进化速率缓慢、序列同源性高,被广泛用于细菌的分类学研究与属种水平的初步筛查[39]。groEL基因是热休克蛋白家族的重要成员之一,广泛存在于病原微生物中,其基因组编码基因在病原菌长期的进化过程中高度保守[40]。目前,E. canis的16S rRNA基因和groEL基因的分子检测技术应用较为成熟,两种基因共同鉴定结果更能说明E. canis的存在。本研究通过对海南黑山羊血液样本开展埃里希体属病原的分子检测,利用16S rRNA基因序列与groEL比对分析的方法,对埃里希体的潜在感染进行分子水平鉴定与验证。该研究旨在通过系统的基因检测手段明确黑山羊体内是否存在E. canis病原,并为海南黑山羊埃里希体病的流行病学调查、风险评估及针对性防控措施制定提供关键的分子依据,填补海南省该领域研究的空白,对保障海南省黑山羊养殖业健康发展具有重要参考价值。
-
2024—2025年,韩谦老师课题组在海南省海口、澄迈、东方、文昌、白沙等多地的黑山羊羊群采集抗凝血样本,共收集1 061份羊血样本,于−80℃保存。
-
仪器名称 型号 公司 高速台式冷冻离心机 Eppendorf 5424R Eppendorf 小型离心机 ABSON MiFly-6 合肥艾本森科学仪器有限公司 小型离心机 Eppendorf 5430 REppendorf 旋涡混合器 QL-901 海门其林贝尔仪器制造有限公司 PCR仪 ABI GeneAmp® 9700 型ABI 测序仪 Illumina Miseq Illumina 电泳仪 DYY-6C 北京市六一仪器厂 全自动凝胶成像分析仪 iBright™ CL750 赛默飞世尔科技公司 移液器 Eppendorf N13462C Eppendorf 超微量分光光度计 NanoDrop2000 Thermo Fisher Scientific Table 1. Main instruments
试剂名称 公司 Blood-DNA Extraction kit 南京诺维赞生物科技股份有限公司 2×TaqPlus-Master-Mix Ⅱ(Dye Plus) DL2000-DNA Marker DL500-DNA Marker 北京宝日医生物技术有限公司 6× Loading Buffer 50×TAE 上海碧云天生物技术股份有限公司 琼脂糖(Agarose) 武汉赛维尔生物科技有限公司 Table 2. Main reagents
-
取冻存抗凝血于冰盒中解冻,参照血液基因组DNA提取试剂盒说明书操作:吸取200 μL 血样至2 mL EP管,加200 μL 缓冲液GB与20 μL Proteinase K,充分颠倒混匀后56℃孵育10 min;室温放置3 min,加350 μL缓冲液BD颠倒混匀,将混合液全部转移至CG2吸附柱,13 000 r·min−1离心1 min,弃废液后将吸附柱放回收集管;向吸附柱加入500 μL缓冲液GDB离心弃废液,再加600 μL 漂洗液PWB离心弃废液并重复1次;13 000 r·min−1离心2 min,弃收集管,室温晾干吸附柱后移入新1.5 mLEP管,加40 μL洗脱缓冲液TB,13 000 r·min−1离心2 min,收集洗脱液于−20℃保存备用。
-
参考文献[31]合成E. canis的16S rRNA基因引物,以及本课题组设计的groEL基因引物。分别用于检测E. canis的16S rRNA基因和groEL基因,引物合成由生工生物工程公司完成。
基因
Gene引物名称
Primer name序列(5′—3′)
Sequence(5′—3′)扩增片段/bp
Product size/bp16S rRNA ECC AGAACGAACGCTGGCGGCAAGC 306 ECB CGTATTACCGCGGCTGCTGGCA ECAN5 CAATTATTTATAGCCTCTGGCTATAGGA 396 HE3 TATAGGTACCGTCATTATCTTCCCTAT groEL groELF ACCTTATGGTGCTCCAGAAATT 772 groELR AGCACCTGTTAAGATAGCAATAT Table 3. Primer sequences
-
以黑山羊全血中提取的基因组DNA为模板,采用PCR技术分别对E. canis的16S rRNA基因和groEL基因进行扩增,并设置阳性对照、无模板对照(ddH2O)和E. canis阴性血液DNA及其他无形体DNA以排除污染,阳性对照来自实验室原有经鉴定过的E. canis阳性样本。16S rRNA基因采用巢氏PCR扩增,第一轮使用引物ECC和ECB,反应体系(25 μL)为2×TaqPlus Master Mix加12.5 μL、上/下游引物(10 μmol·L−1)各1 μL、DNA模板1 μL、ddH2O加9.5 μL;第二轮以第一轮PCR产物为模板加1 uL,引物更换为ECAN5和HE3,反应体系与第一轮一致;groEL基因采用常规PCR扩增,反应体系中DNA模板用量为1 μL,其余体系与16S rRNA的体系相同,所有PCR产物均置于−20℃冰箱保存备用。
-
PCR产物经1%琼脂糖凝胶电泳分离后,通过凝胶成像系统采集图像并记录结果,与标准分子量Marker比对分析后,选取呈阳性条带的产物送至生工生物工程(上海)股份有限公司进行测序鉴定。测序获得的原始序列经下载后,使用Snapgene8.0软件进行处理;处理后的序列上传至NCBI数据库,采用BLAST工具进行序列同源性比对。最后,利用MEGA X软件,以邻接法(Neighbor-Joining,NJ)构建系统发育树(phylogenetic tree)。
-
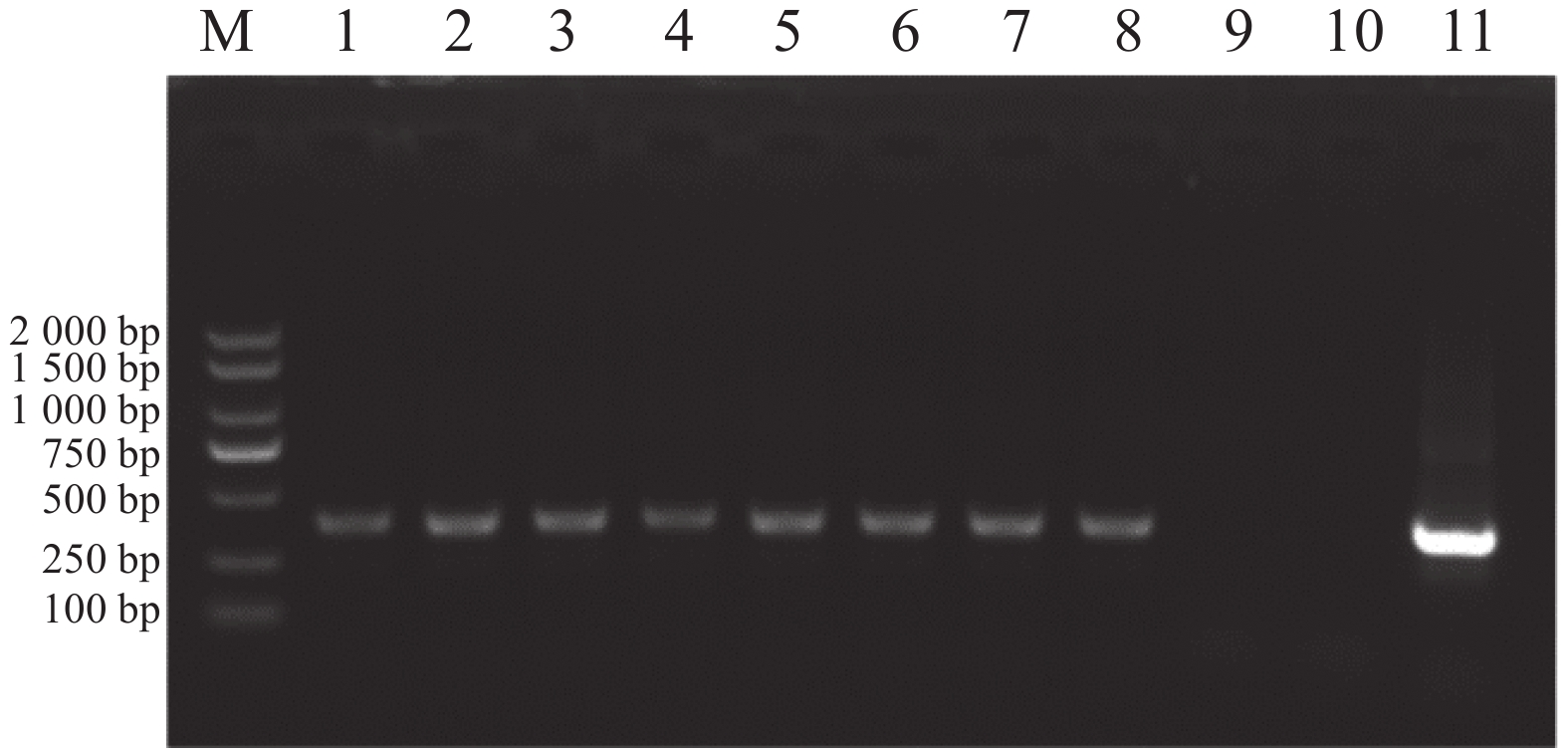
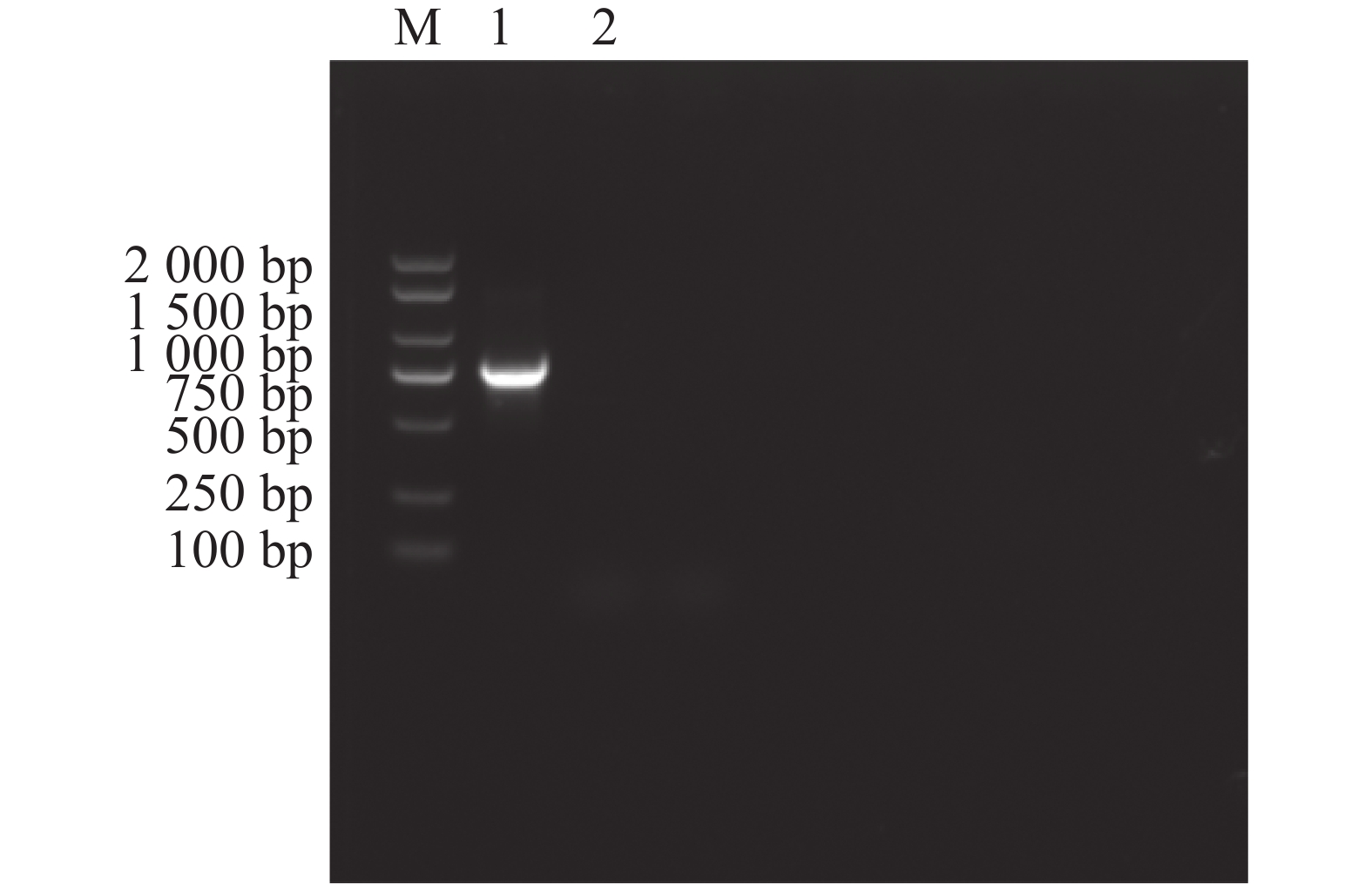
以E. canis的16S rRNA基因和groEL基因为靶标进行PCR扩增后,将产物经1%琼脂糖凝胶电泳分离,通过凝胶成像系统获取图像(图1和图2)。结果显示,图1中16S rRNA基因扩增片段与标准分子量Marker比对后,位于250 bp与500 bp之间且更接近500 bp,图2中groEL基因扩增片段位于750 bp与
1000 bp之间更接近750 bp,两种基因的扩增产物大小均与预期的396 bp和772 bp相符。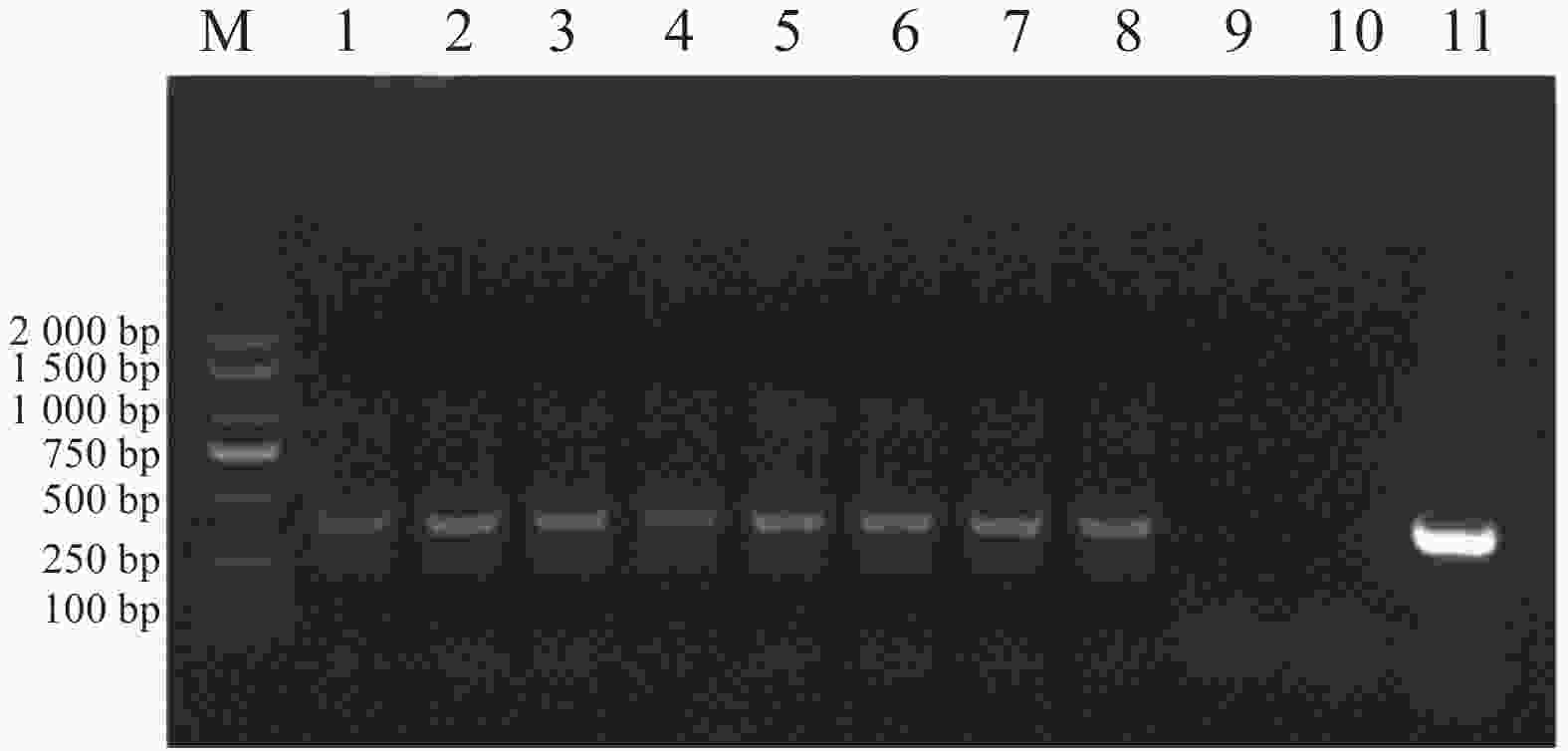
Figure 1. Amplification results of the E. canis 16S rRNA gene.
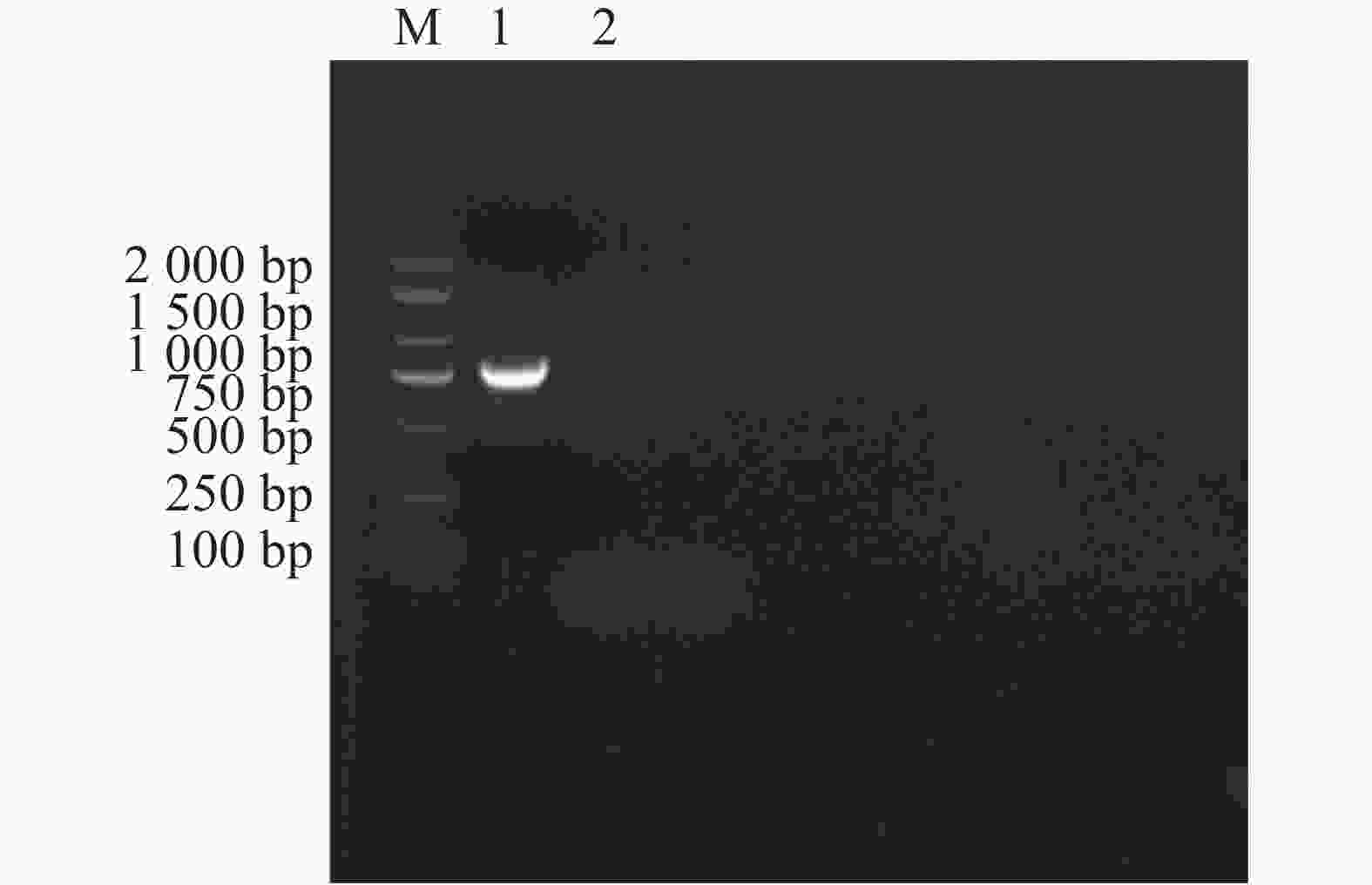
Figure 2. Amplification of the E. canis groEL gene.
-
本次调查共采集海南黑山羊抗凝血样本1 061份,通过PCR方法进行E. canis病原检测,结果显示不同地区羊群中E. canis的感染情况存在差异。在8个采样地区中,文昌、东方及五指山3个地区均未检出E. canis阳性样本,检出率为0;其余5个地区均存在不同程度的感染,其中定安县检出率最高为11.40%(4/35),其次为儋州市7.52%(10/133)和澄迈县2.70%(16/591),白沙县4.00%(2/50)和海口市1.51%(2/132)检出率相对较低。基于样本采集量,澄迈县采集样本数量最多(591份),其阳性检出数量(16份)也为各地区最高,但检出率并非最高。
羊血样本
Sheep blood
sample样本数目/个
Number of
samples/n阳性样本数/个
Number of positive
samples/n检出率/%
Detection
rate澄迈 591 16 2.7 海口 132 2 1.5 定安 35 4 11.4 文昌 54 0 0 儋州 133 10 7.5 白沙 50 2 4 东方 6 0 0 五指山 60 0 0 合计 1 061 34 3.2 Table 4. Detection results of Ehrlichia canis in dogs from various regions of Hainan
-
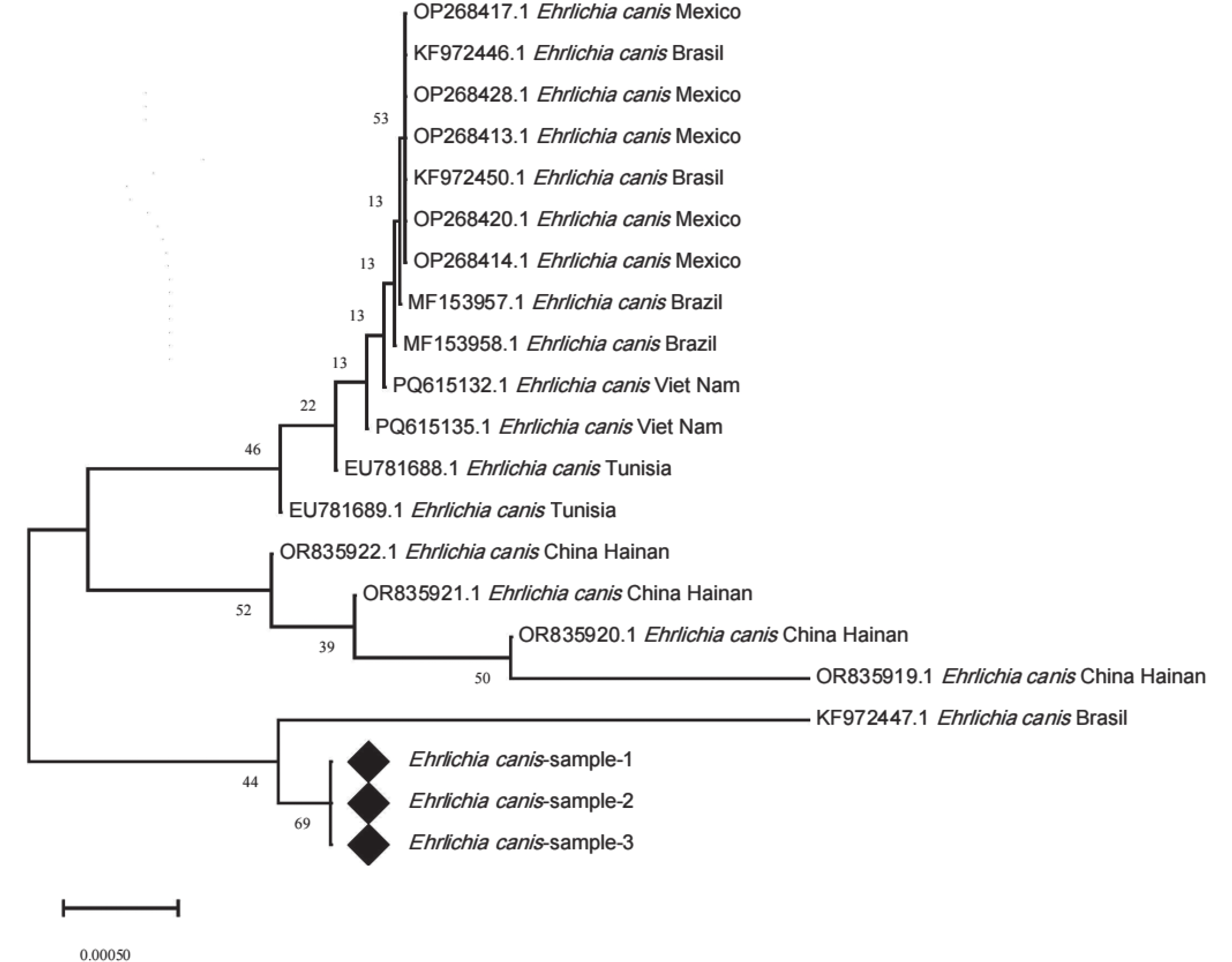
E. canis的16S rRNA基因测序数据进行分析,(通过SnapGene 8.0软件完成序列的剪切、拼接,获得目的序列,将整理后的序列提交至NCBI数据库,利用Nucleotide BLAST工具进行同源性比对),筛选出3条与E. canis同源性较高99.49%的序列,分别命名为Ehrlichia canis-sample-1、Ehrlichia canis-sample-2及Ehrlichia canis-sample-3。从GenBank数据库中下载国内外已报道的多株E. canis的16S rRNA基因参考序列,联合上述3条序列通过MEGA X软件采用MUSCLE算法进行多序列比对,基于比对结果构建系统发育树。本研究所获的3条序列聚为同一分支,并形成高度相似的单系群,表明三者在16S rRNA序列上几乎完全一致。该单系群与巴西提交的序列(KF972447.1)聚为一支,亲缘性最近,同源性99.49%;墨西哥、越南及突尼斯等地区的E. canis序列(OP268417.1、PQ615312.1、EU781688.1)分别形成若干地域性分支,与目的序列偏远较大;中国海南提交的序列(OR835919.1、OR835920.1、OR835921.1及OR835922.1)则构成独立的簇状分支,与样本亲缘性较远,同源性84.95%。上述结果表明,样本与巴西株KF972447的系统发育位置更为接近,与中国海南过去提交的序列分化较大。
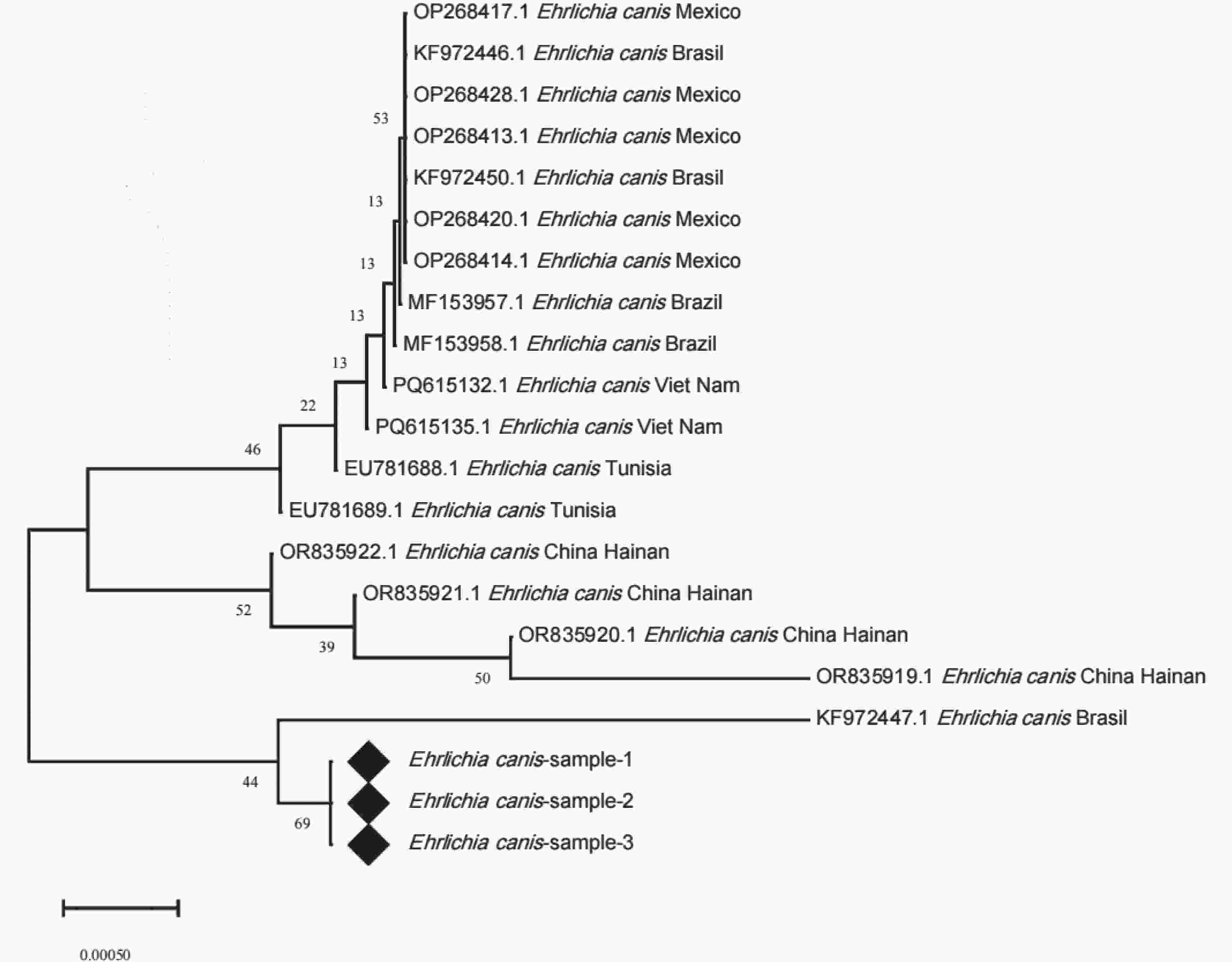
Figure 3. Phylogenetic tree based on the 16S rRNA gene sequences of E. canis
-
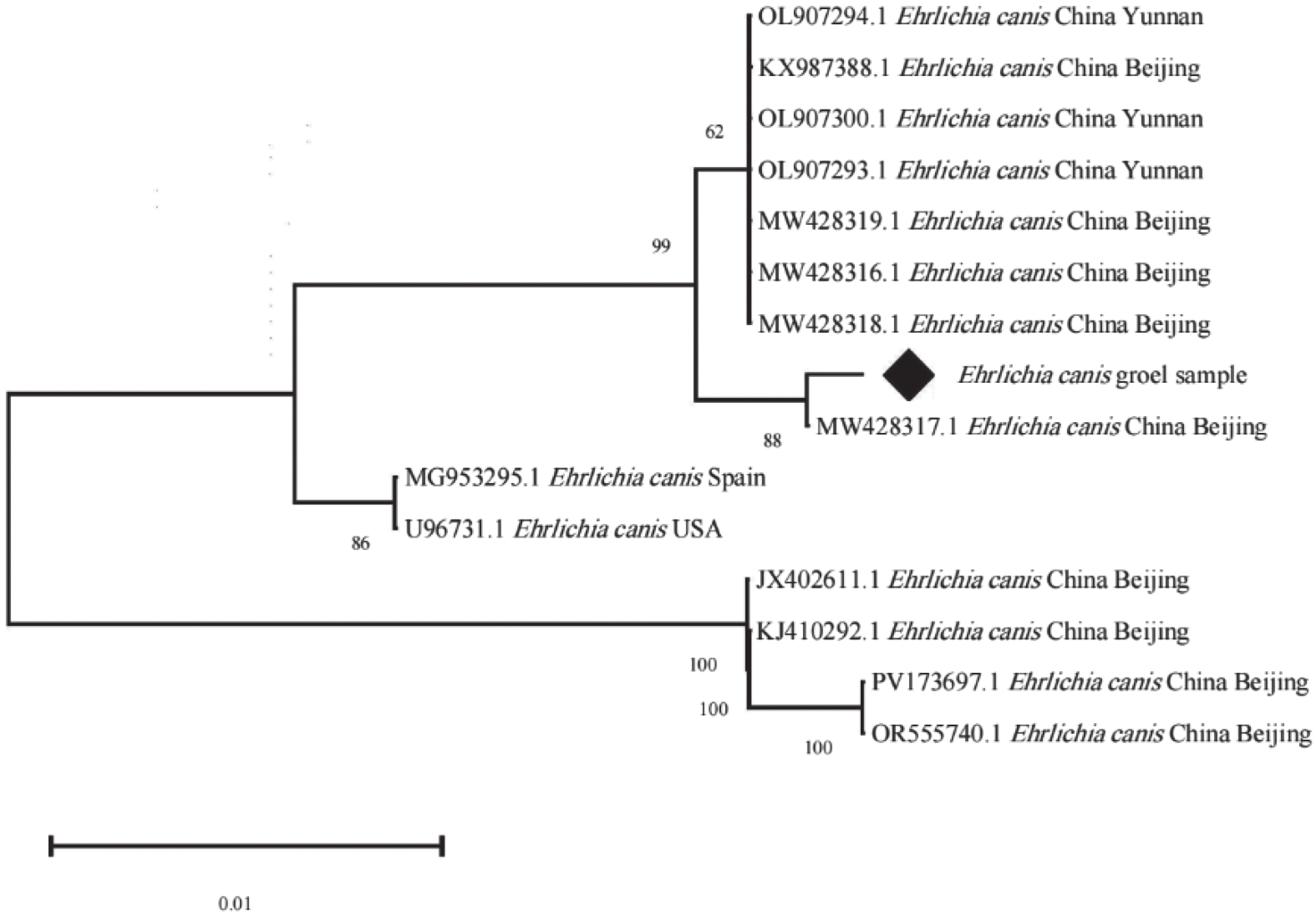
PCR阳性产物由生工生物工程公司测序,将测序得到的序列分别进行剪切、拼接和分析,用MEGA X进行同源性比对,挑选1个序列命名为Ehrlichia canis groEL sample,构建系统树。系统树结果表明,样本与中国北京提供的序列(MW428317.1)聚为一支,亲缘性最近,相似性99.72%。该近缘对与另一较大的中国的北京和云南提交的序列簇(MW428318.1、MW428316.1、MW428319.1 、OL907293.1、OL907300.1、KX987388.1、OL907294.1 等)形成旁系群,亲缘性较近,同源性为99.30%;与中国谱系的其他序列相比,另一明显独立的北京分支(JX402611.1、KJ410292.1、PV173697.1、OR555740.1),同源性95.66%,说明该独立的北京分支与样本所属分支存在明确的谱系分化。
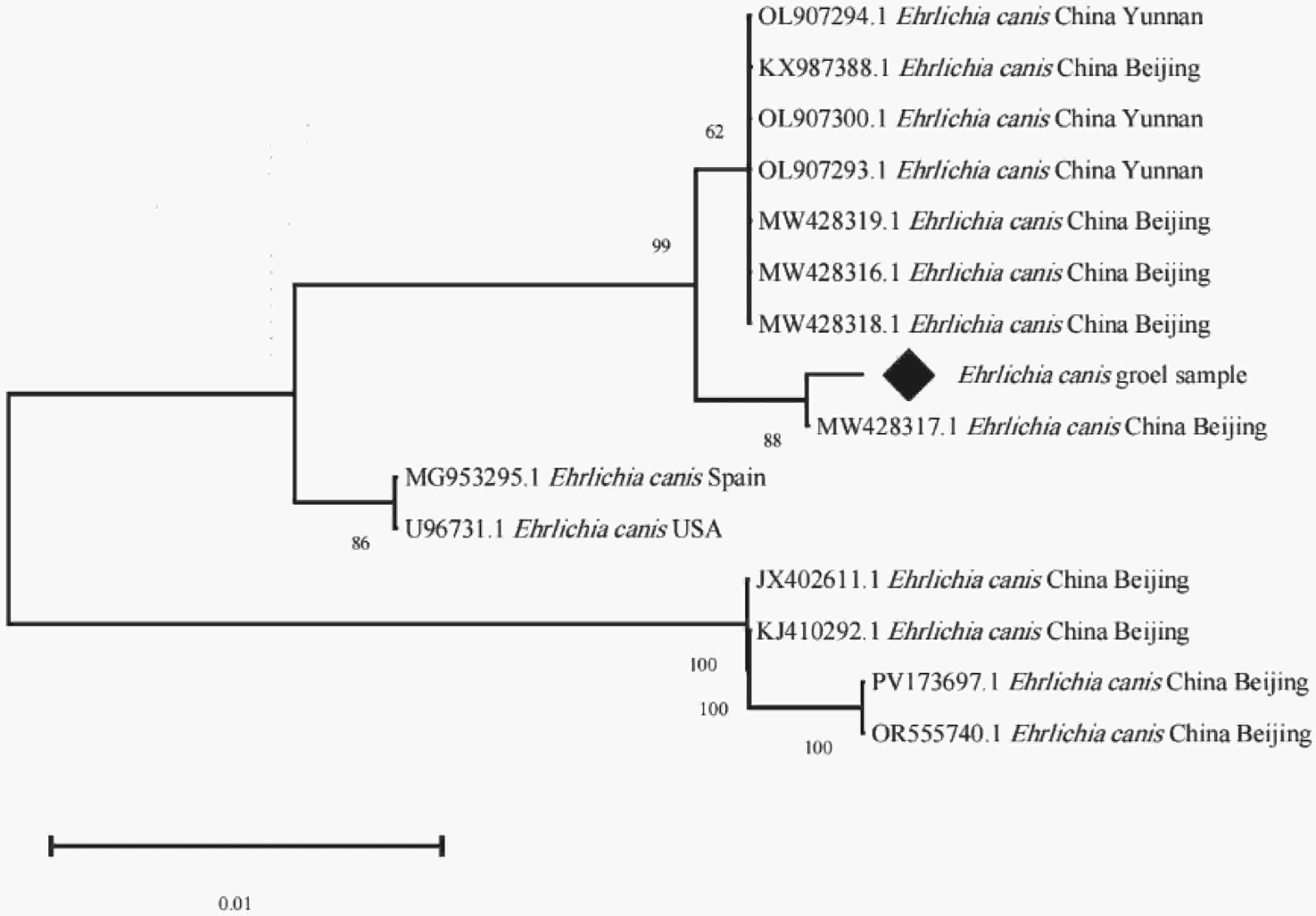
Figure 4. Phylogenetic tree based on the groEL gene sequences of E. canis
-
在欧洲、非洲、美洲等地的非犬科动物检测到E. canis[20-23],在中国的甘肃、贵州等地也发现了反刍动物和鹿的血液中含有E. canis病原体[32-34],这说明非犬科动物可作为该病原体的中间宿主,也表明E. canis的宿主范围突破传统犬类宿主限制。文献[35-37]报道在海南省犬和蜱虫样本中检测到E. canis病原体。探究E. canis在海南省非犬科动物的感染情况,本研究采集了海南黑山羊血液样本,根据PCR扩增结果结合同源比对分析证实在海南黑山羊血样本中首次检测到E. canis,从16S rRNA序列和groEL基因序列的比对分析,证实E. canis能够在黑山羊体内存在。通过对海南省8个地区黑山羊样本的病原检测,检测结果呈现出E. canis在不同地区的检出率差异较大,呈现出区域性流行特征;整体阳性率为3.2%,其中定安县感染率最高11.40%(4/35),儋州市7.52%(10/133)次之,澄迈县2.70%(16/591),这种差异可能与采样地区的宿主、气候、数量等相关。海南地处热带气候,为蜱虫的繁殖与传播提供了优良条件。血红扇头蜱作为E. canis的主要传播媒介,在海南广泛分布[28],且常寄生于犬、羊等动物体表,增加了病原跨宿主传播的风险。此外,黑山羊散养放养的饲养模式使黑山羊与蜱虫的接触机会加大,也为E. canis病原的传播提供了机会。
遗传分析结果显示,海南黑山羊来源的E. canis菌株具有显著的遗传特殊性。基于16S rRNA与groEL基因的系统树分析结果,本研究所有阳性样本聚为单系群,遗传结构高度一致,这可能是由于存在共同的感染源导致的。16S rRNA序列与巴西株(KF972447.1)亲缘性最近,同源性99.49%,而与墨西哥、越南、突尼斯等地分离株分化明显。本研究所获得序列与海南地区过去提交的E. canis菌株序列差异显著,同源性仅约84.95%,形成旁系群,该菌株可能是海南省首次发现或由于外来动物、蜱虫传播导致。groEL序列与中国北京株(MW428317.1)同源性达99.72%,与云南、北京部分序列形成旁系群,说明E. canis种内已出现地区遗传分化。综合分析表明,海南黑山羊E. canis与海南过去提交的序列存在明显进化差异,可能通过外来动物或蜱虫传播进入海南。
从流行病学角度看,在海南黑山羊体内检测到E. canis序列具有重要意义。E. canis感染导致犬出现发热、血小板减少及全血细胞减少等症状,严重时可引发慢性消耗性或致死性疾病[16]。近年来有研究表明,E. canis具有潜在的人畜共患风险[24, 38]。本研究首次在海南发现并证实E. canis存在于反刍动物宿主,揭示E. canis具备跨宿主感染的潜能,宿主范围不断扩大。E. canis可能通过蜱虫在犬与反刍动物之间传播,黑山羊充当中间宿主。本研究检测到的E. canis在遗传上与海南过去菌株差异显著,且可能存在多种遗传型并存,或为外源型分支,也可能经动物调运或蜱虫迁移引入。
综上所述,本研究首次在海南黑山羊中检测并证实E. canis特异性序列,证实本研究检测到的E. canis可感染反刍动物,潜在的跨宿主传播特性,打破了对海南E. canis宿主范围原有的认知。通过16S rRNA与groEL基因的系统树分析,发现本研究菌株与巴西及北京的菌株亲缘性较近,与海南菌株的既往序列差异显著,这可能是由于新出现的或未发现的遗传型导致。本研究为海南E. canis的流行病学监测和防控提供了重要依据。然而,受各地区样本数量限制,结果可能未能展示出E. canis在全省的分布特征。后续拟扩大样本数量和采样范围,结合蜱虫和犬血样本,进一步阐明E. canis的传播机制,为海南省对E. canis的防控提供更系统的数据支持。
Molecular epidemiological investigation of Ehrlichia canis in black goats from Hainan
DOI: 10.15886/j.cnki.rdswxb.20250160
- Received Date: 2025-11-03
- Accepted Date: 2026-01-06
- Rev Recd Date: 2026-01-12
-
Key words:
- Ehrlichia canis /
- Black goat /
- Molecular epidemiology /
- Hainan
Abstract: To investigate the infection status, molecular characteristics, and genetic relationship of Ehrlichia canis in Hainan black goats, a total of 1 061 anti-coagulated blood samples were collected from eight cities/counties (Haikou, Chengmai, Dingan, Baisha, Dongfang, Wenchang, Wuzhishan, Danzhou) of Hainan Province. Detection and phylogenetic analysis were performed by PCR amplification targeting the 16S rRNA and groEL genes of E. canis. The results showed there were 34 positive samples, with an overall positive rate of 3.2%. The samples in Dingan County had the highest detection rate (11.40%), suggesting regional prevalence of the pathogen in this area. Phylogenetic analysis indicated that E. canis strains from Hainan black goats were clustered into the same branch, possibly originating from a common infection source. The 16S rRNA sequences of E. canis from Hainan black goats were most closely related to those of a Brazilian strain (KF972447.1), while the groEL gene showed the highest affinity with a Beijing strain from China (MW428317.1), forming a paralogous group with sequences from Beijing and Yunnan in China, reflecting geographic genetic differentiation. These findings provide the first molecular evidence of E. canis in Hainan black goats, suggesting that E. canis may achieve cross-host transmission via ticks, which offers a reference for further research on the molecular epidemiology and control of E. canis.
| Citation: | Wang Quanjiang, Liu Wenjing, Wang Jiyang, Wu Peifei¹, Yang Haorui, Wang Jinhua. Molecular epidemiological investigation of Ehrlichia canis in black goats from Hainan[J]. Journal of Tropical Biology. doi: 10.15886/j.cnki.rdswxb.20250160

|
 DownLoad:
DownLoad: